Autora : Encarnación Martinez Cazorla
4º Curso de Medicina grupo "C" (curso 2023-24)
Código de trabajo : 2303-EMC
INTRODUCCIÓN
La hemofilia adquirida (HA) es un trastorno hemorrágico autoinmune causado por el desarrollo de autoanticuerpos específicos que inhiben cualquiera de los factores de coagulación. El objetivo más frecuentemente afectado por estos anticuerpos es el factor VIII.
El principal indicio para sospechar la presencia de esta patología es el sangrado agudo en cantidades anormales, y/o en localizaciones infrecuentes en pacientes sin coagulopatía. Suele afectar a pacientes frágiles y se asocia con una alta tasa de mortalidad (9-33%). (1)
En el 48 % de los casos de HA, la enfermedad coexiste con otras patologías:
- Autoinmunes, como lupus eritematoso sistémico, artritis reumatoide, esclerosis múltiple, pénfigo, enfermedad inflamatoria intestinal crónica, asma o reacciones alérgicas graves.
- Linfoproliferativas y tumores sólidos, como linfoma, leucemia, macroglobulinemia, próstata y pulmón.
- Primíparas, en los primeros tres meses de puerperio. (2)
EPIDEMIOLOGÍA
La HA presenta una incidencia muy baja. Alrededor de 1.3 a 1.5 casos/millón de personas/año, aunque muy probablemente muchos casos no llegan a diagnosticarse. (2) Estas cifras realmente son poco representativas debido a la falta de registros y lo difícil del diagnóstico cuando no hay sospecha clínica.
Suele aparecer de media a los 65 años, pero cuenta con dos picos de edad más frecuentes. Un primer pico en mujeres jóvenes a partir del posparto o en presencia de enfermedades del colágeno; y el segundo pico afecta a mayores de 60 años sin grandes diferencias de género. (1)
A continuación, observamos un gráfico bimodal de incidencia, el que se aprecia como ya hemos comentado, un pico de mujeres fértiles entre 19 y 40 años; y un segundo en adultos mayores (mediana de edad 73.9 años), con un ligerísimo predominio en varones (1.4:1). (2)

Figura 1. Distribución de casos de hemofilia adquirida del registro europeo EACH2. (2)
FISIOPATOLOGÍA
Inmunológicamente hablando la HA va a producirse a raíz de la producción de IgG1 e IgG4, policlonales, de cadenas ligeras kappa, contra “hot spots” de los dominios A1, A2 y C2 del FVIII.
Realmente se puede producir autoanticuerpos contra cualquier factor hemostático, pero las características del FVIII hacen que sea el blanco más idóneo.
Se cree que la pérdida de tolerancia al FVIII se debe a factores genéticos y ambientales. La predisposición genética se basa en la asociación entre HA y polimorfismos del antígeno 4 de linfocitos T citotóxicos y expresión de los alelos DRB1*16 y DQB1*050210 del HLA clase II
Además, se produce un desequilibrio de las células TCD4 + Th1 y Th2, lo que permite generar el autoanticuerpo. Si predomina la subclase de IgG inducida por células T CD4-Th2 se producirán concentraciones mayores de autoanticuerpo, lo cual supone un peor pronóstico; por el contrario, si predominan las Th1, habrá una mejor respuesta al tratamiento inmunosupresor.
Finalmente, los autoanticuerpos asociados con HA inhiben la función del FVIII o aumentan su depuración, cayendo así el efecto hemostático. Al reaccionar contra el FVIII inhiben su interacción con el FIX activado y con el FX, alterando la generación de trombina y fibrina (2).
CUADRO CLÍNICO
En la HA es importante el diagnóstico temprano que permita tratar la hemorragia evitando procedimientos invasivos peligrosos, ya que, por lo general, más del 90% de los episodios de sangrado al diagnóstico son graves y requieren internación, por lo que buscamos reducir esta cifra. El diagnóstico tardío aumentará la mortalidad de esta afección, para favorecer el diagnóstico precoz es mejor contar con un hematólogo con experiencia en inhibidores y un laboratorio especializado en hemostasia.
El sangrado típico de la HA suele ser extenso y afectar a piel, mucosas y tejido celular subcutáneo en forma de equimosis o hematomas muy característicos que, cuando ocurren en los miembros inferiores o superiores, pueden producir un síndrome compartimental por isquemia secundaria a compresión de tejidos. Otra posibilidad, es que aparezca en forma de hematomas musculares espontáneos o ante traumas mínimos, o como una hemorragia digestiva, pulmonar, sangrado post parto o del sistema nervioso central.
Suele estar acompañado de anemia y no presenta hemartrosis. En ocasiones, el sangrado puede ser interno, por lo que hay que monitorizar con pruebas de imagen. (3)

Fig. 2. Hematoma en flanco y dorso en paciente con hemofilia adquirida. (3)
Cuando el inhibidor se asocia al embarazo aparecerá entre 2 y 5 meses post parto, sin síntomas durante el embarazo normalmente. Lo más frecuente es que ocurra en primíparas, sin reaparición en los embarazos siguientes. (3)
DIAGNÓSTICO DE LABORATORIO
En un paciente sano, con autoanticuerpos contra FVIII, encontraremos un aPTT prolongado, un TP normal y un tiempo de trombina normal.
En el caso de encontrar un aPTT prolongado aislado hay varias opciones, puede deberse a la presencia de un inhibidor lúpico, un inhibidor especifico de un factor, heparina o a un déficit de factores de la coagulación.
El inhibidor lúpico debe descartarse realizando pruebas específicas. La presencia de heparina se detecta a través de un TT alterado.
Si a los resultados el aPTT resulta prolongado se hará un estudio de mezclas para distinguir entre inhibidores y falta o déficit de algún factor puntual de la cascada de coagulación.
El inhibidor de FVIII es tiempo y temperatura dependiente, por lo que el aPTT de la mezcla se prolongará aún más si se lo incuba. En los pacientes con hemofilia adquirida el nivel plasmático de FVIII estará disminuido o ausente.
El método Bethesda será el empleado para cuantificar el inhibidor específico de FVIII una vez detectado, mide la potencia del anticuerpo. (3)
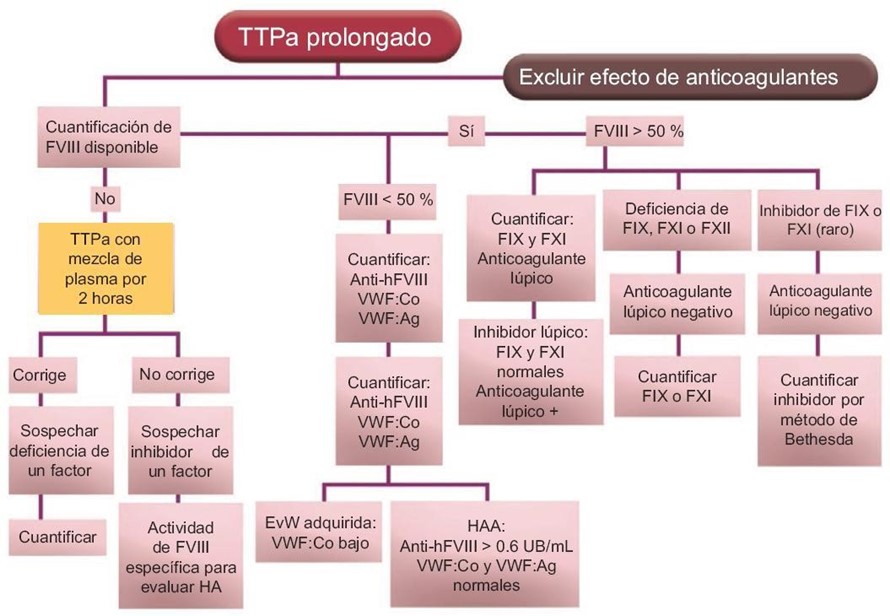
Figura 3. Manejo del paciente con el tiempo de tromboplastina parcial activada prolongado. (2)
COMPLICACIONES Y PRONÓSTICO
El diagnóstico precoz mejora el pronóstico en gran medida. Este va a depender de la gravedad de la hemorragia y la enfermedad subyacente.
Hay casos en los que la enfermedad puede remitir de forma espontánea (25- 36%), cuando está asociada con embarazo y toma de medicamentos. Por lo general, presenta una supervivencia del 69-78% y, una vez que remite, será similar a la de la población general. Un 8% de los afectados sufrirán una hemorragia incontrolable que les causará una muerte temprana. Otra posibilidad es la muerte temprana por hemorragia gastrointestinal o pulmonar. La hemorragia intracraneal y retroperitoneal se asocia con mortalidad más tardía.
La muerte del paciente puede ser debido a la HA, la enfermedad concomitante o los efectos secundarios de la enfermedad primaria. También hay un alto índice de mortalidad debido al tratamiento de la enfermedad primaria. La HA con mejor supervivencia es la asociada al embarazo.
Hay una serie de factores de mal pronóstico para remisión y supervivencia:
- FVIII:C <1 UI/dL.
- Escala funcional de la OMS >2.
- Asociado con neoplasias.
- No remisión completa.
- Edad >65 años.
Mientras que los factores de buen pronóstico son:
- Remisión completa.
- Actividad del FVIII >1 UI/dL.
- Título de inhibidor <16 UB.
Si se logra remitir la enfermedad hay que llevar acabo un monitoreo clínico y por laboratorio debido al riesgo de recurrencia, ya que hasta el 20 % recae en los dos primeros años. Se realizarán revisiones mensuales los primeros seis meses, cada dos o tres meses los siguientes seis meses y cada seis meses durante el seguimiento por dos años. Para la HA asociada con el embarazo, la recurrencia en embarazos subsecuentes es baja. La recaída se manejará con el tratamiento inicial (2).
TRATAMIENTO
El tratamiento de la HA tiene dos objetivos:
- Controlar la hemorragia si está presente o prevenir su aparición.
- Erradicar el inhibidor.
El tratamiento debe instaurarse rápidamente ya que estos pacientes pueden llegar a presentar una alta mortalidad en caso contrario. Están contraindicados los procedimientos invasivos a menos que sea estrictamente necesario, y en caso de que no se pueda postergar instaurar tratamiento hemostático previo, esta opción reduce las hemorragias iatrógenas.
También es importante la prevención de traumatismos por parte del paciente y evitar analgésicos no esteroideos, así como informar al paciente de los síntomas tempranos de hemorragia para que pueda estar alerta. Que el paciente sea cociente de la importancia de buscar atención en caso de hemorragia no justificada.
No todos los pacientes necesitan tratamiento hemostático al diagnóstico, un ejemplo es si presenta equimosis extensas. Se tratará de erradicar el inhibidor de inmediato. Lo mismo ocurre con los pacientes con inhibidor y deficiencia funcional del FVIII:C. sin hemorragia.
Los enfermos que recibirán tratamiento hemostático inmediato son los que presenten riesgo hemorrágico alto o hemorragia clínica: muscular, genitourinaria, gastrointestinal, retroperitoneal, articular, pulmonar o del sistema nervioso central. Recibiendo este grupo tratamiento hemostático antes y después de someterse a un procedimiento invasivo mayor.
El tratamiento inicial de la hemorragia grave es con agentes de puenteo.
El CCPa contiene los factores II, VII, IX y Xa que puentean al FVIII para generar trombina. En pacientes con hemorragias crónicas y leves se usa profilácticamente la mitad de la dosis. El FVII activado recombinante (rFVIIa) permite puentear la hemostasia desde la vía extrínseca, evitando a la intrínseca. La evidencia del rFVIIa como primera línea de tratamiento en HA es grande, su eficacia llega al 100 %. La efectividad y seguridad es discretamente mayor con rFVIIa que con CCPa, pero mayor con ambos que con FVIII o desmopresina. No hay estudios que hagan preferible a uno u otro. Aunque se desconoce la duración óptima del tratamiento, puede mantenerse hasta controlar la hemorragia, y se recomiendan dosis adicionales para prevenir su reaparición. Finalmente, rFVIIa y CCPa pueden administrarse alternadamente en hemorragias graves resistentes si la monoterapia se administra a dosis y frecuencia máximas (2).
CONCLUSIÓN
El mayor problema con la HA es el diagnóstico tardío. Son muy importantes las estrategias de prevención, así como la educación de la población y de los médicos para concienciar de la enfermedad y aprender a identificarla, ya que una vez incluida en el diagnóstico diferencial, las pruebas para confirmarla son bastante sensibles. Otro problema es la falta de registros epidemiológicos. En cuento al tratamiento, la gran fragilidad del paciente, inmunodeprimido, supone un reto, por elevado riesgo de sepsis.
BIBLIOGRAFÍA
1.- Hemofilia adquirida: epidemiología, presentación clínica, diagnóstico y tratamiento. Medicina Clínica (Edición inglesa), Volumen 148, Número 7, 7 de abril de 2017, páginas 314-322.
2.- Gac. Méd. Méx vol.156 no.1 Ciudad de México ene./feb. 2020 Epub 26-Mayo- 2021.
3.- Medicina (B. Aires) vol.75 no.4 Ciudad Autónoma de Buenos Aires ago. 2015.
No hay comentarios:
Publicar un comentario
Se admiten comentarios que indiquen posibles errores en los textos y/o sugerencias de temas, y/o propuestas de mejoras en el blog y/o dudas sobre la realización de trabajos. Dado que este blog no es un consultorio médico, no se responderán preguntas realizadas con esa finalidad. Los comentarios que se consideren inapropiados, serán eliminados de inmediato, sin ningún tipo de excepción.
Nota: solo los miembros de este blog pueden publicar comentarios.