Autora : Ana Buedo Macarro
4º Curso de Medicina grupo "A" (curso 2024/25)
Código de trabajo : 2407-ABM
INTRODUCCIÓN
La Leucemia Eritroide Aguda (LEA) es un subtipo raro pero agresivo de Leucemia Mieloide Aguda (LMA) y constituye el 2% de todos los casos de LMA. Suele estar asociada con un cariotipo complejo, así como con la pérdida bialélica de TP53.
La mayoría de los casos de LEA surgen de novo, aunque un subgrupo surge a partir de un síndrome mielodisplásico (SMD) previo o de otras neoplasias mieloides crónicas. No existen asociaciones definitivas de factores genéticos y ambientales con el desarrollo de la LEA (1).
Desde el primer reconocimiento de una malignidad hematológica predominantemente eritroide a principios del siglo XX, la LEA ha pasado por una serie de cambios en sus definiciones y nomenclatura, incluyendo eritoleucemia, mielosis eritremica, LMA-M6 y leucemia eritroide pura (2).
La biología agresiva de esta enfermedad y sus características moleculares, la convierten en difícil de manejar y curar, siendo la supervivencia media de entre 3 a 9 meses (1).
Actualmente, los enfoques de tratamiento son ineficaces y señalan la urgente necesidad de terapias novedosas.
HISTORIA Y DEFINICIONES
La primera descripción de una leucemia predominantemente eritroide fue de Giovanni Di Guglielmo cuando en 1917, publicó un informe sobre eritroleucemia, que detallaba un caso de leucemia caracterizado por la proliferación tanto leucocítica como eritrocítica.
Acuñó posteriormente el término "mielosis eritremica" en 1923 para describir un trastorno sanguíneo agudo y proliferativo que involucraba solamente eritrocitos nucleados. En un editorial publicado en Blood en 1958, se caracterizó el síndrome de Di Guglielmo como una enfermedad trifásica, primero con predominancia eritroblástica, luego progresando a través de una fase mixta leucoblástica/eritroblástica y finalizando con leucemia mieloblástica. Ahora se entiende que la LEA se describe más precisamente con la definición de mielosis eritremica y que la progresión a un fenotipo mieloblástico no se observa con frecuencia.
La clasificación Franco-Americana-Británica (FAB) de leucemias agudas en 1976, reconoció la leucemia eritroide como un subtipo de LMA, denominándola específicamente LMA-M6. LMA-M6 se definió como aquella con >30% de mieloblastos en la celularidad de la médula ósea con ≥10% de precursores eritroides mostrando displasia, análoga a la LMA definida actualmente como relacionada con mielodisplasia. Esta clasificación fue revisada en 1985, y la definición de LMA-M6 cambió para requerir que ≥ 50% de la celularidad de la médula fuera de células eritroides nucleadas mientras que ≥ 30% de las células mieloides no eritroides fueran mieloblastos.
Aun así, algunos pensaron que la definición de LMA-M6 era demasiado amplia y no diferenciaba entre el síndrome de Di Guglielmo, en el cual los elementos inmaduros son predominantemente mieloblastos, y la enfermedad de Di Guglielmo, en la cual son predominantemente proeritroblastos. Para abordar esto, demostraron que los pacientes con LMA-M6 y predominancia de mieloblastos tenían resultados significativamente mejores en comparación con aquellos con predominancia de proeritroblastos. Propusieron dividir LMA-M6 en M6a, correspondiente a la categoría tradicional FAB M6 y M6b, correspondiente a una leucemia eritroide pura con > 30% de proeritroblastos.
En 2001, la Organización Mundial de la Salud (OMS) publicó su clasificación inicial de neoplasias mieloides, con criterios del grupo FAB. En su 3ª clasificación, la OMS reconoció 2 subtipos de LEA basados en el tamaño de la población de mieloblastos.
- El primer tipo, leucemia eritroide/mieloide aguda (correspondiente a FAB LMA-M6), se definió como ≥ 50% de precursores eritroides en la población celular nucleada de la médula y mieloblastos ≥ 20% de la población celular no eritroide.
- El segundo tipo, denominado leucemia eritroide pura (M6b), se definió por ≥ 80% de precursores eritroides inmaduros con mínima diferenciación y sin población significativa de mieloblastos.
Los autores de esta clasificación mencionaron que la leucemia eritroide pura se había referido previamente como enfermedad de Di Guglielmo. La 4ª edición de OMS no introdujo cambios importantes en la definición de LEA, solo señalando que algunos casos con displasia significativa (≥ 50% de células en ≥ 2 linajes) y mieloblastos ≥ 20% deberían ser categorizados como LMA con cambios relacionados con mielodisplasia, independientemente de la población de precursores eritroides.
Las revisiones de la 4ª edición cambiaron el denominador utilizado para calcular el porcentaje de mieloblastos de “células no eritroides” a todas las células nucleadas de la médula ósea, reclasificando una gran proporción de LEA (subtipo eritroide/mieloide aguda) como neoplasia mielodisplásica. Los casos restantes de LEA (subtipo eritroide/mieloide aguda) fueron reclasificados, dejando la leucemia eritroide pura como la única leucemia eritroblástica verdadera. La definición de leucemia eritroide pura se mantuvo en gran medida sin cambios respecto a la original, solo especificando la presencia de ≥ 30% de proeritroblastos dentro del ≥ 80% de precursores eritroides inmaduros en la médula ósea.
Por último, la 5ª edición de la clasificación de la OMS (2022), cambió la nomenclatura de la leucemia eritroide pura a LEA, y dejó la definición sin cambios mientras destacaba la importancia de las mutaciones TP53 en la patogénesis de la enfermedad. La evolución de la nomenclatura y los criterios diagnósticos se puede encontrar en la Tabla 1 (2).
Tabla 1. Evolución de los criterios diagnósticos y de
la nomenclatura de las neoplasias mieloides predominantemente eritroides (2).
Click para agrandar la imagen.
EPIDEMIOLOGÍA
La edad media de diagnóstico de la LEA es de 67 años, aunque hay estudios que han demostrado una edad bimodal de diagnóstico con un pequeño pico alrededor de los 20 años y un segundo pico más grande en los primeros 70 años. Esta enfermedad también muestra un leve predominio masculino sobre femenino (2.4:1) (1).
ETIOLOGÍA
La mayoría de los casos de LEA se desarrollan de novo, representando aproximadamente el 1% de todas las LMA de novo, y la enfermedad no está asociada a ningún factor de riesgo identificable. Se han descrito casos raros de eritroleucemia familiar de novo, autosómica dominante con penetrancia variable. En la literatura se han descrito casos de LEA que evolucionan a partir de otras enfermedades antecedentes como SMD (anemia refractaria con exceso de blastos o citopenia refractaria con displasia multilinaje), neoplasias mieloproliferativas (leucemia mieloide crónica con crisis eritroblástica) y exposición a toxinas como el benceno, denominándose LEA secundaria. Esta también se ha descrito en pacientes con antecedentes de otros tipos de cáncer tratados con quimioterapia, tratamiento inmunosupresor o radiación ionizante (3).
La presentación clínica de la LEA puede ser inespecífica, ya que los síntomas y hallazgos más prominentes al diagnóstico son fiebre y palidez, anemia (hemoglobina media de 7.5 g/L), hepatoesplenomegalia y evidencia de eritropoyesis ineficaz (que puede asemejarse a la hemólisis).
La presentación extramedular como un sarcoma mieloide es inusual en pacientes con LEA, pero puede afectar a localizaciones extramedulares.
Se deben verificar antecedentes de SMD, neoplasias mieloproliferativas y niveles de eritropoyetina, ya que esta información influye en la clasificación diagnóstica (1).
Los pacientes pueden ser sintomáticos durante un tiempo antes del diagnóstico, estableciéndose este entre 1 a 3 meses. Es raro que los pacientes con LEA sean sintomáticos durante más de 6 meses antes del diagnóstico inicial. También hemos observado pacientes que, aunque sintomáticos, eran muy funcionales en el momento del diagnóstico, lo que llevó a los clínicos a dudar del diagnóstico y a considerar un tratamiento de prueba para la deficiencia nutricional (3).
DIAGNÓSTICO
El diagnóstico, al igual que en otros tipos de leucemias agudas se basa en la demostración de las células leucémicas en médula ósea, sangre periférica o tejidos extramedulares, aunque los estudios han planteado la preocupación por la calidad subóptima de las biopsias en la LEA, así como la frecuente falta de blastos en sangre periférica, lo que hace el diagnóstico algo más difícil.
Las características más típicas en la biopsia de médula ósea son la hipercelularidad, la diseritropoyesis y un alto porcentaje de precursores eritroides (Imagen 1). Las características frecuentemente observadas en el frotis de sangre periférica de pacientes con LEA son leucopenia, estiramiento basofílico y morfología anormal de los glóbulos rojos; sin embargo, estas no son características diagnósticas ni distintivas de la LEA (1).
Imagen 1. Cambios morfológicamente representativos en médula ósea de un paciente con leucemia eritroide acuda, subtipo eritroleucémico. En A y B se observa hiperplasia eritroide, aumento de blastos, diseritropoyesis y sideroblastos en anillo (3).
Hallazgos inmunohistoquímicos y por citometría de flujo
Los marcadores de inmunohistoquímica y citometría de flujo se utilizan para caracterizar mejor los casos de LEA:
CD71 es el receptor de transferrina en la superficie celular y está presente en la mayoría de los precursores eritroides y suele estar sobreexpresado en los blastos de LEA y precursores malignos eritroides.
Los blastos inmaduros en la LEA pueden expresar antígenos del grupo sanguíneo Gerbich (Gerb), E-cadherina, anhidrasa carbónica 1, antígenos CD36 y CD68 también. Así mismo, hay tenue expresión de hemoglobina y de la glicoproteína-A.
Otros marcadores, como GLUT1, han mostrado teñirse positivamente en células de la línea eritroide (1).
La mieloperoxidasa (MPO) y HLA-DR son típicamente negativas en citometría de flujo, lo que puede ayudar a distinguir entre LMA y LEA (2).
Hallazgos citogenéticos
Un cariotipo complejo se define como al menos tres anomalías citogenéticas y es una característica casi uniforme de la LEA. En el contexto de esta patología, las deleciones en 5q y 7q, la monosomía 5 y 7, y la trisomía 8 son las anomalías más detectadas. Además, una anomalía cariotípica que puede estar presente en el cromosoma 17 (17p13) se ha asociado con la pérdida de función de p53 descrita en un porcentaje significativo de pacientes con LEA.
Se ha descrito variación citogenética entre pacientes con LEA cuando se estratifica por edad. Un estudio que se centró en las características citogenéticas de la LEA examinó a 31 pacientes y encontró que los menores de 45 años tenían más anomalías citogenéticas en comparación con los mayores de 45 años (66.7% vs. 54.5%).
Varios estudios concluyen que las anomalías citogenéticas t(8;21), t(15;17), inv(16) y del(20q), se asocian con mejores resultados entre los pacientes con LEA. En cuanto a los peores, se asocian con las anomalías −5, −7 y 3q.
La mayoría de los estudios concluyen que la ausencia de cariotipo complejo, aunque sea muy rara en la LEA, podría estar asociada con mejores resultados y una supervivencia más prolongada (1).
Hallazgos moleculares
Por lo que respecta a la patobiología (Imagen 2), se han explorado varios mecanismos implicados basados en datos procedentes de muestras humanas y modelos murinos, exponiendo a continuación algunos de ellos:
- Activación del Receptor de Eritropoyetina (EPOR) y la Vía JAK2: se ha demostrado cierta relación directa entre la activación del receptor de eritropoyetina y la patogénesis de la LEA, ya que parece estar sobreexpresado.
- GATA Binding Protein 1 (GATA1): GATA1 tiene un importante papel en la eritropoyesis. Se han identificado cambios en la transcripción de GATA1 en el 25% de pacientes con LEA, que reducen su expresión o la expresión ectópica de sus interactores ERG y SPI1, disminuyendo la accesibilidad de la cromatina en sitios de unión de GATA1 y promoviendo fenotipos leucémicos (1).
- Factores de transcripción ETS (ERG, SPI y FLI1): ERG coopera con GATA1 para controlar la diferenciación eritromegacariocítica y se ha relacionado con un pronóstico desfavorable en LMA. SPI1 y FLI1 inhiben la diferenciación eritroide, y la sobreexpresión de estos factores en modelos murinos induce fenotipos de eritroleucemia, convergiendo en el GATA1 como el principal desencadenante del desarrollo de la patología.
- Expresión de Oncogenes Maestros: Los proto-oncogenes como c-MYC están sobreexpresados en modelos murinos, inhibiéndose la diferenciación eritroide y promoviendo la eritroleucemia. Las mutaciones en los oncogenes H-Ras y K-Ras se asocian con la transformación leucémica en LEA.
- Actividad TP53 dañada: la LEA tiene alta prevalencia de mutaciones bialélicas de TP53, más frecuentes en casos secundarios. A pesar de que su implicación funcional en la diferenciación eritroide no está completamente clara, se sabe que cooperan con otras vías para promover la proliferación de células madre hematopoyéticas. Coexisten con otras alteraciones genéticas como mutaciones en KRAS, NRAS, BCOR y otros genes de señalización y reparación de ADN. Se impulsa así la eritroleucemia, demostrándose esto en modelos murinos, donde la pérdida de TP53 junto con mutaciones en BCOR y otros genes, generan tumores agresivos (1).
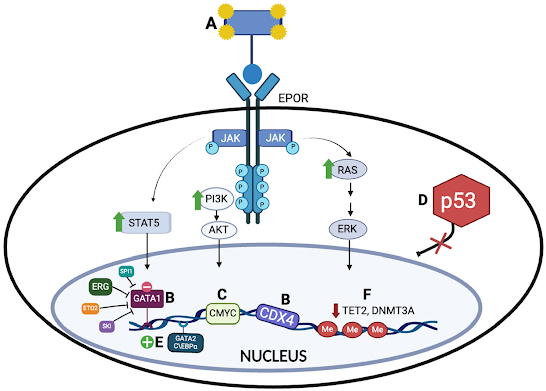
Imagen 2. Mecanismos implicados en la patobiología de la leucemia eritroide aguda (LEA)
DIAGNÓSTICO DIFERENCIAL
Se incluyen tanto patologías neoplásicas como benignas.
Para el subtipo de eritroleucemia, las entidades más importantes en el diagnóstico diferencial incluyen SMD con predominancia eritroide, LMA con cambios relacionados con displasia mieloide y otros tipos de LMA con aumento de precursores eritroides. Las características más importantes para distinguir en el diagnóstico diferencial son el porcentaje de blastos y la presencia de displasia multilineal (2 o 3 linajes) en al menos el 50% de las células de un linaje dado.
Los pacientes con SMD suelen tener anemia, y en la médula ósea suele observarse predominancia eritroide, reflejando un intento de compensar la anemia. Morfológicamente, la displasia eritroide y los sideroblastos en anillo se observan tanto en SMD como en el subtipo de eritroleucemia. El factor clave que ayuda a distinguir es el porcentaje de blastos en médula ósea. Si el recuento de blastos es inferior al 20% de todas las células nucleadas y también inferior al 20% de los elementos no eritroides, el diagnóstico de SMD será más apropiado.
La leucemia mieloide aguda con cambios relacionados con la displasia mieloide se caracteriza por un 20% o más de blastos y displasia multilineal en al menos el 50% de las células en dos o más linajes. Estos casos pueden presentar 50% o más de precursores eritroides, pero no se clasifican como LEA, según los criterios actuales de la OMS. Además, los casos con al menos un 20% de blastos en sangre periférica o médula ósea y anormalidades citogenéticas relacionadas con SMD o un historial previo de SMD, incluso cuando están asociados con una población de precursores eritroides en la médula ósea del 50% o más, deben considerarse casos de este tipo de LMA.
Otros tipos de LMA también pueden asociarse a un aumento en el número de precursores eritroides cuando se diagnostican inicialmente. En todos estos casos, los blastos representan un 20% o más de todas las células nucleadas de la médula ósea. Cuando los precursores eritroides representan el 50% o más de las células nucleadas, según nuestra experiencia, la explicación más común es la terapia previa con eritropoyetina (EPO), que aumenta los precursores eritroides, causando diseritropoyesis. Es por eso que, en el contexto de la terapia con EPO, es difícil establecer el diagnóstico de LEA.
Se revisaron gran número de casos, pertenecientes al Centro Oncológico MD Anderson de la Universidad de Texas (Houston), en los que predominaban los precursores eritroides y se habían diagnosticado como LEA, observándose que un subconjunto de ellos tenía un historial de terapia con EPO. Así mismo, la quimioterapia previa también puede resultar en una eritrocitosis de rebote (3).
Ahora, refiriéndonos al subtipo de leucemia eritroide pura se incluye la leucemia megacarioblástica aguda, leucemia linfoblástica aguda (LLA), linfomas y trastornos no neoplásicos. Los casos del subtipo de leucemia eritroide pura en los que hay evidencia clara de maduración eritroide son más fáciles de reconocer. Sin embargo, también hay casos de este subtipo en los que se observa maduración eritroide mínima o nula y están compuestos por blastos inmaduros. Para estos, se necesitan análisis inmunofenotípicos y citogenéticos.
La leucemia megacarioblástica aguda puede ser más difícil de diferenciar de la leucemia eritroide pura. En la citogenética, la tinción MPO es negativa en ambas y el PAS muestra positividad citoplasmática en ambas. Emplearemos entonces análisis inmunofenotípico. Los casos de leucemia eritroide pura expresan uno o más antígenos asociados a los eritroides (como la hemoglobina A o la glucoforina), mientras que los casos de leucemia megacarioblástica aguda expresan antígenos asociados a megacariocitos como CD41, CD61 y el factor VIII. Sin embargo, muchos de estos pueden estar ausentes y, parece haber casos de leucemia eritroide pura con expresión de CD41 o CD61. No está claro si estos casos deben clasificarse entonces como leucemia mixta aguda eritroide-megacarioblástica. La microscopía electrónica para mostrar la peroxidasa de plaquetas puede ser útil para diagnosticar leucemia megacarioblástica aguda, así como la presencia de la anomalía t(1;22)(p13;q13) y las anormalidades que afectan al cromosoma 3q26.
Los casos de leucemia linfoblástica aguda (LLA) y linfomas, expresan antígenos de células B o T, y la LLA suele ser positiva para la transferasa terminal deoxinucleotidil (TdT), siendo el inmunofenotipado útil para distinguir estas entidades de la leucemia eritroide pura.
Varias enfermedades no neoplásicas pueden causar predominancia eritroide en médula ósea, y por lo tanto deben ser excluidas. Estas incluyen, anemia megaloblástica debido a deficiencia de vitamina B12 o ácido fólico, intoxicación por metales pesados, efectos de medicamentos y, diseritropoyesis congénita. Con un historial completo y pruebas de laboratorio pertinentes podremos descartar estas posibilidades (3).
TRATAMIENTO
Debido a la rareza del diagnóstico de LEA y la escasa comprensión de la biología de la enfermedad, las opciones de tratamiento son limitadas. Como primera línea se incluye quimioterapia intensiva y agentes hipometilantes (HMA). El trasplante de médula ósea alogénico (alo-TMO) es el único enfoque potencialmente curativo, pero requiere previa remisión completa.
Ǫuimioterapia Intensiva
Se utiliza como tratamiento de primera línea en pacientes aptos para terapia intensiva. En un gran estudio retrospectivo multinacional de 217 pacientes con LEA realizado en 2017, 122 pacientes fueron tratados con quimioterapia, empleándose un régimen de inducción con daunorubicina (45 o 60 mg/m² × 3 días) y citarabina (100 mg/m² bid × 7 días). Otros regímenes incluyeron idarubicina (12 mg/m²) o mitoxantrona (12 mg/m²) en combinación con citarabina. El grupo sometido a quimioterapia tenía una edad media de 60 años al diagnóstico, en comparación con 69 para el grupo tratado con HMA. La tasa de respuesta objetiva fue del 72%, ocurriendo respuesta completa en 79 pacientes (66%), parcial en 7 (6%), enfermedad estable en 16 (13%) y, progresión primaria de la enfermedad en 17 (14%). Tras el tratamiento con quimioterapia, 23 (18.8%) recibieron un alo-TMO, experimentando una mediana de supervivencia global de 5.9 meses.
Este régimen de tratamiento a una edad más joven también se ha asociado con una supervivencia global significativamente mayor. Un análisis retrospectivo reciente de 2023, demostró una relación inversa entre la edad y la supervivencia global en pacientes con AEL, con una mediana 69, 18, 8, 3 y 1 mes para los grupos de edad <18, 18–49, 50–64, 65–79 y >80, respectivamente. La LEA pediátrica muestra un perfil genómico distinto, lo que podría explicar los resultados más favorables a esta edad. Además, el régimen de quimioterapia pediátrica es más agresivo, ya que los niños lo toleran mejor que los adultos.
A pesar de que el efecto de la quimioterapia es más pronunciado en pacientes pediátricos, su uso está asociado a una mayor supervivencia tanto en poblaciones pediátricas como en adultas, demostrando una diferencia significativa en la supervivencia global entre los pacientes que recibieron este tratamiento y aquellos que no (1).
Agentes Hipometilantes (HMA)
Azacitidina y decitabina se utilizan en pacientes con LMA no aptos para quimioterapia intensiva. Los HMA funcionan revirtiendo la metilación del ADN que a menudo silencia los genes supresores de tumores involucrados en la señalización de la patogénesis del cáncer. Aunque pueden inducir una respuesta inicial prometedora en un subconjunto de pacientes con LEA, casi todos los pacientes suelen recidivar. Esto puede ocurrir a través de recidiva primaria, en la que no hay mejora después de cuatro a seis ciclos de HMA, o secundaria, en la que los pacientes progresan tras una respuesta inicial.
Sin embargo, un estudio retrospectivo reciente de 41 pacientes con LEA describió que no había beneficio al usar un régimen de tratamiento sobre otro. Los regímenes reportados en este estudio incluyeron monoterapia con HMA, HMA junto con venetoclax y quimioterapia intensiva. Por lo tanto, se necesitan más estudios para comprender mejor los diversos grados de eficacia entre los tratamientos para LEA según los criterios actualizados de 2022.
Venetoclax El venetoclax es un inhibidor selectivo de la proteína BCL-2, que ha mejorado las tasas de remisión y la superviviencia global de pacientes con LMA no aptos para quimioterapia. Sin embargo, los subtipos de LMA eritroide/megacariocítico se asocian a resistencia a este fármaco. Esto se debe a la diferenciación eritroide/megacariocítica, además de a las mutaciones TP53. La pérdida de p53 se ha vinculado con la resistencia al venetoclax, junto con una regulación compensatoria concomitante de BCL-XL. De hecho, se ha observado que las células de LMA que exhiben diferenciación eritroide/megacariocítica dependen de BCL-XL en lugar de BCL-2 para su supervivencia, de forma que la inhibición de BCL- 2 tiene efecto terapéutico limitado en LEA (1).
Trasplante de Médula Ósea Alogénico (alo-TMO)
Como se ha mencionado con anterioridad, es el único enfoque potencialmente curativo, pero requiere remisión completa previa, que rara vez se logra. Se ha encontrado previamente que la supervivencia media de los pacientes con LEA que recibieron alo-TMO fue de 66 meses desde el trasplante. Sin embargo, la definición de LEA para este análisis se basó en la clasificación de la OMS de 2008. La mediana de supervivencia global de los receptores de alo-TMO fue de 89 meses, en comparación con 5 meses para aquellos que no se sometieron. Por lo tanto, esta terapia de consolidación puede mejorar los resultados en LEA (1).
Terapia con Células T con Receptor de Antígeno Ǫuimérico
Esta terapia ha comenzado a usarse en leucemias y linfomas al dirigirse a antígenos distintos presentes en células malignas. Recientemente, algunos estudios han examinado los posibles resultados de la terapia CAR-T en LMA. En un estudio de 2023, se investigaron datos de secuenciación de ARN de células individuales de individuos con LMA y tejido sano para determinar epítopos expresados selectivamente en células malignas. A través del análisis computacional y la validación posterior, los autores encontraron que el receptor de factor estimulante de colonias 1 y el clúster de diferenciación 86 podrían usarse como objetivos para esta terapia. Así mismo, en 2019, se demostró que CD7 expresado por blastos malignos y células progenitoras de un subconjunto de pacientes con LMA, servía como objetivo para la terapia CAR-T. En general, la variedad de epítopos moleculares que se han identificado en la terapia CAR-T para LMA podría representar aplicaciones futuras beneficiosas para el tratamiento.
Aunque hay avances prometedores en la terapia con células CAR-T para el tratamiento de algunas leucemias, incluida LMA, los estudios que investigan el análisis de células individuales en LEA siguen siendo limitados. Dado que hay pocas opciones de tratamiento, la terapia CAR-T debe ser investigada más a fondo como una modalidad de tratamiento (1).
Direcciones Futuras
El tratamiento de la leucemia eritroide aguda (LEA) continúa siendo un desafío ante los escasos resultados clínicos y la falta de ensayos clínicos específicos. A pesar de los avances en el trasplante alogénico de médula ósea (alo-TMO) y nuevas terapias dirigidas, las tasas de supervivencia en los últimos 20 años no han mejorado.
Uno de los principales retos es que los factores transcripcionales que impulsan la LEA no son fácilmente tratables, y la inhibición de JAK2 no controla la enfermedad.
Sin embargo, estudios recientes han demostrado sensibilidad a la inhibición de CDK7, CDK9 y PARP en modelos de LEA mutados en genes como TP53, BCOR, y DNMT3A, lo que sugiere que estas terapias podrían llegar a ser efectivas. La combinación de decitabina con inhibidores de PARP, por ejemplo, podría amplificar los efectos citotóxicos en casos de LEA mutados en TP53.
Además, el inhibidor BCL-XL se ha demostrado como objetivo terapéutico, particularmente en leucemias eritroide y megacarioblástica. Al combinar inhibidores de BCL-XL con ruxolitinib (inhibidor de JAK2) se observó respuesta sinérgica en líneas celulares y modelos de xenoinjertos. Así mismo, se ha encontrado eficacia con el inhibidor navitoclax, que apunta a BCL-XL y BCL-2, respaldándose así el uso de inhibidores de BCL-XL como futura estrategia de tratamiento para la LEA.
Estudios recientes también sugieren que la inhibición de JAK2 es efectiva en pacientes con LEA que presentan mutaciones en TP53 y alteraciones en la señalización de JAK2/EPOR.
Se estan llevando a cabo ensayos clínicos que incluyen el uso de talazoparib o gemtuzumab ozogamicina, demostrándose remisión completa en pacientes con LMA refractaria, pero se necesitan más estudios para confirmar los hallazgos preclínicos y optimizar los tratamientos (1).
CONCLUSIONES
La leucemia eritroide aguda es un subtipo altamente agresivo y raro de LMA caracterizado por una citogenética compleja de alto riesgo, mutaciones frecuentes en TP53 y una predominancia de precursores eritroides y proeritroblastos en la médula ósea. Está asociado a malos resultados clínicos y, la nomenclatura vaga y cambiante ha obstaculizado su reconocimiento.
Su diagnóstico es establecido mediante biopsia de médula ósea, citometría de flujo e inmunohistoquímica, los cuales confirman la presencia de los precursores eritroides y las características de la enfermedad.
Debido a sus características de alto riesgo, los resultados clínicos siguen siendo pobres. Los agentes hipometilantes (HMA), la quimioterapia intensiva y el trasplante alogénico de medula ósea (alo-TMO) se han utilizado con eficacia general limitada. La edad más joven y el uso de quimioterapia ayudan en el tiempo de supervivencia; sin embargo, el alo-TMO es el único enfoque potencialmente curativo (1).
En conclusión, para mejorar los resultados, debe haber un enfoque concertado y colaborativo en la investigación, además de mayor desarrollo terapéutico, para evitar que continúe siendo una patología cuyo diagnóstico es en gran medida intratable.
BIBLIOGRAFÍA
1.- Johns Hopkins School of Medicine, B. M. (06 de Junio de 2024). Europe PMC. Obtenido de Sitio web de Europe PMC : https://europepmc.org/article/MED/38892446
2.- Department of Hematology and Oncology, M. U. (28 de Abril de 2023). ScienceDirect . Obtenido de Sitio web de ScienceDirect: https://
www.sciencedirect.com/science/article/pii/S2152265023001398?vi a%3Dihub
3.- Zhuang Zuo, M. P., Jacek M. Polski, M., Armen Kasyan, M., & L. Je|rey Medeiros, M. (01 de Septiembre de 2010). Allen Press. Obtenido de Sitio web de Allen Press : https://meridian.allenpress.com/aplm/article/134/9/1261/461132/Acute- Erythroid-Leukemia










