Autora : Irene Pinar Sánchez
4º Curso de Medicina grupo B (curso 2022/23)
Código de trabajo : 2205-IPS
INTRODUCCIÓN
La leucemia promielocítica aguda (LPA) es un subtipo (M3) de leucemia mieloide aguda (LMA). No obstante, la LPA presenta particularidades biológicas y clínicas que la diferencian del resto de LMAs.
Se caracteriza por un comportamiento clínico agresivo cuyo curso, en ausencia de tratamiento, es fatal Sin embargo, el descubrimiento de los detalles moleculares de su patogénesis, posibilitó avances en su tratamiento y en particular, la introducción de medicamentos diferenciadores como el ácido todo transretinoico (ATRA) y, más recientemente, el trióxido de arsénico (ATO) que han hecho que el pronóstico de esta leucemia haya mejorado de una manera importante.
De modo que, hoy en día, la leucemia promielocítica se considera una enfermedad curable con tasas de supervivencia superiores al 90 % a los 2 años de seguimiento (1).
HISTORIA DE LA LEUCEMIA PROMIELOCÍTICA AGUDA
La primera descripción de la enfermedad fue publicada en 1957 por Leif Hillestad, quien señaló que podría tratarse de la forma más maligna de leucemia (2).
Los tratamientos iniciales de inducción incluyeron agentes quimioterápicos como 6- mercaptopurina en monoterapia o en combinación con esteroides, metil-glioxalguanil hidracina, metotrexato y posteriormente arabinósido de citosina y antraciclinas, que, si bien en algunos casos fueron efectivos, sobre todo estas últimas, condujeron de forma general a resultados desfavorables de manera específica en las respuestas a largo plazo.
Un gran avance en el conocimiento de la enfermedad ocurrió en 1978, cuando se descubrió que las células leucémicas podrían sufrir diferenciación bajo la influencia de varios agentes. A principios de 1980 se experimentó con una amplia variedad de compuestos que incluyeron ácido retinoico y se demostró que inducía maduración morfológica y funcional en cultivos celulares con detención en su diferenciación y una respuesta específica de las muestras de LPA.
En 1985 se consideró utilizar el ATRA (ácido transretinoico), un isómero del ácido retinoico, que demostró ser superior a otros isómeros, tanto en entornos in vitro como in vivo, para inducir la diferenciación terminal en células LPA frescas.
Pero no fue hasta 1990 cuando se descubrió la base molecular de la respuesta al ATRA. El punto de ruptura 17q21 se ubicó dentro del locus del gen del receptor alfa del ácido retinoico y el punto de ruptura del cromosoma 15q22 en el gen de la leucemia promielocítica. Luego, se demostró que este gen se involucra en el control de la proliferación celular, apoptosis y senescencia. En tanto, la clonación de la translocación t (15;17) a partir de la reacción en cadena de la polimerasa con transcripción inversa constituyó el inicio de estrategias para el diagnóstico rápido y detección sensible de la enfermedad mínima residual. Así, la persistencia o reaparición de positividad de RT-PCR para PML-RARα en pacientes con remisión morfológica fue correlacionado con la inminente recaída hematológica (2).
El ATRA producía un cambio conformacional en la proteína quimérica PML/ RARα, lo que daba lugar a la reactivación de la transcripción génica inicialmente reprimida por el reclutamiento de factores correpresores y la hipermetilación de genes promotores. Otro efecto atribuido al ATRA que se descubrió fue la inducción de la degradación de la oncoproteina PML/RARα a partir de la activación de la escisión mediada por caspasas y el sistema ubiquitina-proteasoma.
Asimismo, se vio que el arsénico también inducía la degradación del PML / PML- RARα a través de la producción de especies reactivas de oxígeno, introduciéndose también junto con el ATRA en el tratamiento de la leucemia promielocítica.
Estudios recientes, basados en modelos murinos, demostraron que la combinación de los dos agentes diferenciadores (ATRA + arsénico) prolonga la supervivencia y erradican la enfermedad, a partir del sinergismo de sus efectos terapéuticos.
De este modo, se demostró la posible curación de los pacientes con este tipo de leucemia sin quimioterapia, así como cuestionó la terapia de mantenimiento en pacientes de riesgo bajo e intermedio de recaídas.
EPIDEMIOLOGÍA
La LPA representa hasta el 20 % de todos los casos de LMA, afecta por igual a ambos sexos, se presenta en adolescentes y adultos desde la segunda década de la vida y es rara después de los 60 años. Así como con otras neoplasias de la línea mieloide, se ha observado un aumento del riesgo de LPA en las personas expuestas a agentes quimioterapéuticos, especialmente a las antraciclinas y el etopósido (1).
ETIOLOGÍA
Como hemos comentado antes, la mayoría de los casos de LPA son causados por un reordenamiento del cromosoma 17 con el cromosoma 15: t(15;17). Pueden darse casos de reordenamientos del cromosoma 17 con otros cromosomas, pero son muy raros.
Este reordenamiento se da a un nivel específico del cromosoma 15, en la región donde se encuentra el gen PML, el cual quedará truncado y en cuanto al cromosoma 17 se dará en la región del gen RARα (receptor del ácido retinoico). Se hereda, pues, un gen de fusión PML/RARα y su contrapartida el oncogén RARα/PML, con su derivada oncoproteína (3).
Hay una proporción de los enfermos que tienen también una o dos anormalidades adicionales (trisomía 8: t (15;17)+8; anormalidades del cromosoma 7: t (15:17) abn (7) ...) lo que provoca un impacto negativo en los enfermos.
MANIFESTACIONES CLÍNICAS
Los individuos con LPA pueden tener manifestaciones clínicas similares a las de otros pacientes con LMA, en relación con los síntomas de la anemia, el sangrado asociado a trombocitopenia e infecciones secundarias a la disminución del recuento de neutrófilos. No obstante, lo que hace particularmente especial a este tipo de leucemia desde el punto de vista clínico, y lo que le confiere la alta mortalidad temprana, es el desarrollo de una coagulopatía grave. Esta está presente, usualmente, en el momento del diagnóstico y sin manejo adecuado tiene un curso fatal.
La coagulopatía en la LPA es de origen multifactorial, participan elementos tales como la trombocitopenia asociada con la infiltración medular, la coagulación intravascular diseminada asociada con niveles aumentados de factor tisular, y un estado de hiperfibrinólisis primaria (1). Por esta razón, las personas con LPA pueden cursar de manera temprana con sangrado grave que afecta, en especial, al sistema nervioso central (SNC), los pulmones y, en menor medida, al tracto gastrointestinal y las superficies mucosas.
A pesar de que la coagulopatía hemorrágica es el evento más frecuente y temido de la enfermedad al momento del diagnóstico, se pueden también presentar complicaciones trombóticas hasta en el 10 % de los casos.
VARIANTES
A parte de la leucemia promielocítica típica, hay otra variante:
- Variante microgranular M3: en contraste con la M3 Hipergranular clásica, la cifra de leucocitos en la M3 microgranular suele estar notablemente aumentada. La célula predominante en la sangre periférica es un promielocito con núcleo bilobular, reniforme o multilobulado y citoplasma desprovisto de gránulos o con sólo unos cuantos azurófilos. La cromatina nuclear es fina y frecuentemente con nucléolos visibles. En mayor parte de los casos está presente como una población celular menor, un promielocito anormal con núcleo bilobulado, citoplasma basófilo intenso y a veces prolongaciones citoplásmicas, pero en ocasiones ésta puede ser la célula predominante. Cuando hay prolongaciones citoplásmicas, las células semejan megacarioblastos (4).

Figura 1: Diagnóstico diferencial entre la leucemia aguda promielocítica clásica (M3) y la variante (M3v)
DIAGNÓSTICO
El diagnóstico, al igual que en los otros tipo de leucemias agudas se basa en la demostración de las células leucémicas en médula ósea, sangre periférica o tejidos extramedulares. Pero en este caso, la identificación de la t(15;17)(q24;q22) mediante citogenética o del gen de fusión PML-RARα mediante PCR es indispensable para el diagnóstico certero de la LPM en más del 95 % de los casos (2).
Citología:
- M3 típica: promielocitos atípicos con bastones de Auer en empalizada e hipergranulación.
- M3 variante o microgranular: signo del hachazo.
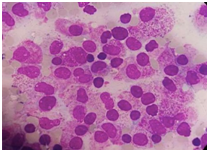
Figura 2: frotis de sangre periférica de un paciente con leucemia aguda promielocítica que muestra promielocitos hipergranulares (5).
Citometría: HLA-DR negativo y MPO positiva 100%.
Coagulación: CID (hipofibrinogenemia con aumento de dímero D y alargamiento de los tiempos de coagulación).
Citogenética y FISH:
- t(15; 17)
- Otras como t(5; 17) y t(11;17)
Biología molecular:
- PML/RARα.
- PLZ/RARα y NPM/RARα.
TRATAMIENTO
El tratamiento de la leucemia promielocítica se centra en la destrucción de la oncoproteína resultante de la fusión de los genes PML/RARα, ya que sin la oncoproteína la célula neoplásica muere. La administración de trióxido de arsénico (ATO) ha demostrado ser muy efectiva en la eliminación de las células neoplásicas ya que se une a la oncopreteína con mucha especificidad e induce su eliminación por la vía del proteasoma (6).
Este se da junto con Ácido Transretinoico (ATRA) a altas dosis para promover la acción fisiológica del receptor de ácido retinoico (prodiferenciativo y promadurativo), ayudando a las células neoplásicas a terminar su diferenciación y maduración.
COMPLICACIONES DEL TRATAMIENTO
La mayoría de los efectos secundarios son temporales y se resuelven una vez completado el tratamiento. Sin embargo, los pacientes con leucemia promielocítica aguda pueden necesitar formas específicas de atención de apoyo. Algunas complicaciones poco frecuentes que pueden aparecer son las siguientes (7):
- Síndrome de diferenciación. El tratamiento de la LPA suele asociarse con una variedad de síntomas y afecciones anormales, entre ellos la retención de líquidos, el esfuerzo para respirar, la acumulación de fluidos cerca del corazón o los pulmones y los episodios de baja presión arterial. Este grupo de síntomas se conoce como “síndrome de diferenciación”. A menudo el síndrome de diferenciación ocurre durante las primeras dos semanas después de iniciar el tratamiento, pero se puede presentar más tarde. Se produce porque al afectar a las células leucémicas con el tratamiento, se liberan muy rápido cantidades grandes de citocinas (sustancias inmunitarias) que se encontraban en el interior de las células causando fiebre, tos, problemas para respirar; aumento de peso; hinchazón de los brazos, las piernas o el cuello, acumulación de líquido alrededor del corazón y los pulmones, presión sanguínea baja e insuficiencia de los riñones. Si no se trata rápido el síndrome de diferenciación es potencialmente mortal. Se debe realizar un estrecho seguimiento de los pacientes para detectar la evolución de estos síntomas, ya que el síndrome de diferenciación junto con la hemorragia son las principales causas de fallecimiento durante la terapia de inducción. La detección temprana y el inicio inmediato de un tratamiento con corticoesteroides son esenciales para manejar esta posible complicación.
- Prolongación del intervalo QT. El uso de trióxido de arsénico puede afectar los niveles de electrolitos (minerales esenciales en la sangre, como potasio, magnesio y calcio), lo que puede causar un trastorno en el ritmo cardíaco conocido como “prolongación del intervalo QT”. Este trastorno causa un ritmo cardíaco acelerado que puede provocar desmayos repentinos o convulsiones. Deben controlarse los electrolitos antes y durante el tratamiento de la leucemia promielocítica aguda para asegurarse de que se mantienen dentro de un rango de referencia normal. Para evitarlo se pueden pedir análisis de sangre de rutina y electrocardiogramas para poder detectar cualquier efecto negativo del trióxido de arsénico.
PRONÓSTICO
El pronóstico de la LPA es muy bueno si se trata correctamente. El desarrollo de nuevas terapias dirigidas a la oncoproteína específica de esta leucemia (PML/RARα) hace que el tratamiento sea muy efectivo y específico contra las células neoplásicas. La administración de sales de arsénico juntamente con ácido retinoico confiere a estos pacientes un pronóstico casi igual a la esperanza de vida de la población general con la edad del paciente, ya que es capaz de eliminar la gran mayoría de células neoplásicas en un período de seis meses. Es una de las pocas leucemias con tratamiento molecular específico eficaz. (2)
CONCLUSIONES
Como resumen, diremos que gracias a ciertos avances científico-técnicos en las últimas décadas del pasado siglo esta variedad de leucemia pasó de ser una de las leucemias agudas más agresivas y de peor pronóstico por su alta mortalidad, a ser la leucemia mieloide de más fácil manejo y de mayor porcentaje de curación al lograrse, en un período relativamente corto, la remisión hematológica y molecular de los enfermos. Todo ello gracias a la introducción de las técnicas de biología molecular y de terapias novedosas, como los inductores de la diferenciación-maduración y de la apoptosis celular (8). Siendo esta, el único tipo de leucemia cuyo tratamiento es el ácido transretinoico.
BIBLIOGRAFÍA
1.- Mejía-Buriticá L, Torres-Hernández JD, Vásquez G de J, Mejía-Buriticá L, Torres- Hernández JD, Vásquez G de J. Leucemia promielocítica aguda. Estado del arte. Iatreia. 2021 Mar;34(1):42–53.
2.- Barroso Sánchez G, Hernández Padrón C, Barroso Sánchez G, Hernández Padrón
C. Terapias dirigidas a dianas moleculares: perspectiva actual en la leucemia promielocítica. Revista Cubana de Hematología, Inmunología y Hemoterapia [Internet]. 2021 Sep [cited 2023 Mar 11];37(3).
3.- Amor Vigil AM, Lavaut Sánchez K, Díaz Alonso CA, Amor Vigil AM, Lavaut Sánchez K, Díaz Alonso CA. Técnicas de citogenética y biología molecular para el diagnóstico y seguimiento de la leucemia promielocítica. Revista Cubana de Hematología, Inmunología y Hemoterapia [Internet]. 2021 Sep [cited 2023 Mar 11];37(3).
4.- Larquin Comet JI, Leyva Diviú A, León Ramentol C, García Fontes Y. Leucemia promielocítica aguda. Comportamiento clínico. Revista Archivo Médico de Camagüey. 2008 Dec;12(6):0–0.
5.- Franco, R. J. V. (2016). LEUCEMIA PROMIELOCITICA AGUDA DE ALTO RIESGO, ASOCIADA A HEMORRAGIA RETINIANA. Revista Médica Carrionica, 3(3).
6.- Miguel JFS, Sánchez-Guijo F. Hematología. Manual Básico Razonado - 5a Edición. 2020. 336 p.
7.- Información sobre la leucemia promielocítica (N26-S) www.LLS.org/espanol
8.- Padrón CH. Avances en el diagnóstico y tratamiento de la leucemia promielocítica en Cuba. Rev Cubana Hematol Inmunol Hemoter. 2014;30(1):1–3.
No hay comentarios:
Publicar un comentario
Se admiten comentarios que indiquen posibles errores en los textos y/o sugerencias de temas, y/o propuestas de mejoras en el blog y/o dudas sobre la realización de trabajos. Dado que este blog no es un consultorio médico, no se responderán preguntas realizadas con esa finalidad. Los comentarios que se consideren inapropiados, serán eliminados de inmediato, sin ningún tipo de excepción.
Nota: solo los miembros de este blog pueden publicar comentarios.