4º Curso Medicina grupo B (curso 2021-2022)
Código de trabajo : 2114-ERA

Ilustración 1: Fisiopatología de la anemia de Blackfan-Diamond. Tomado de : Da Costa, Lydie et al. “Anemia de Diamond-Blackfan”. Blood 136,11 (2020): 1262-1273. doi:10.1182/blood.2019000947. (2)
INTRODUCCIÓN
La anemia de Blackfan-Diamond constituye un síndrome de insuficiencia medular hereditario constitucional que, por lo general, es diagnosticado en el primer año de vida. Es un tipo de anemia congénita, caracterizada por aplasia pura de la serie roja y asociada a anomalías óseas. Exactamente es una anemia macro o normocítica crónica. “Fue identificada por primera vez en 2005 y constituyó la primera ribosomopatía humana” (2). Su principal característica es la eritroblastopenia, relacionada con la variación alélica heterocigota en 1 de los 20 genes de proteínas ribosómicas de la subunidad grande o pequeña. La peculiaridad más importante de la Anemia de Blackfan-Diamond clásica es un defecto en la maduración del ARN ribosómico que genera un estrés nucleolar. Esto conduce a la estabilización del gen p53 y la activación de sus objetivos, lo que genera una detección del ciclo celular y la consiguiente apoptosis. Por otro lado, también hay pacientes que presentan esta eritroblastopenia como resultado de mutaciones en los genes GATA1/HSP70, un desequilibrio de globina/grupo hemo, que junto con un exceso del hemo libre tóxico, va a conducir a la producción de especies reactivas de oxígeno, explicándose así la eritropoyesis defectuosa. Sin embargo, a pesar del conocimiento de toda la fisiopatología y bases genéticas que determinan esta enfermedad, las opciones terapéuticas siguen siendo muy limitadas. No obstante, “los últimos avances en terapias génicas, trasplantes de células madre y nuevos fármacos prometedores, suponen un futuro esperanzador para los pacientes que sufren de esta enfermedad” (2)
Ilustración 1: Fisiopatología de la anemia de Blackfan-Diamond. Tomado de : Da Costa, Lydie et al. “Anemia de Diamond-Blackfan”. Blood 136,11 (2020): 1262-1273. doi:10.1182/blood.2019000947. (2)
EPIDEMIOLOGÍA
Al ser una enfermedad congénita rara, la incidencia es de 7/1.000.000 de nacidos vivos. La incidencia es similar entre diferentes etnias y géneros. El diagnóstico se establece entre los 2 y 3 meses de edad, con el 95% de los casos diagnosticados antes de los 2 años y el 99% antes de los 5. Sin embargo, los avances en el diagnóstico molecular permiten detectarla en pacientes con formas de presentación más atípicas e incluso en adultos con anemia leve.
FISIOPATOLOGÍA
La fisiopatología de esta enfermedad aún no está definida por completo. Cada día se produce en el organismo 200.000.000.000 de glóbulos rojos y debido a que los genes de RP están principalmente involucrados en la traducción del ARNm, se puede plantear la hipótesis de que la traducción de algunos genes específicos podría verse afectada por un defecto en la formación del complejo ribosomal. De todos los genes de proteínas ribosómicas, RPS19 es el gen mutado con más frecuencia, esta mutación da como resultado una deficiencia de la proteína RPS19, conocida como “haploinsuficiencia”. Esta proteína juega un papel importante en la maduración del ARNr 18S y en la síntesis de 40S, por lo tanto, la mutación va a provocar una disminución de la producción de estas subunidades ribosómicas con la consecuente reducción del inicio de la traducción. Esto provoca un impacto negativo en la diferenciación y producción de células progenitoras hematopoyéticas normales y por tanto un incremento de la apoptosis. “La deficiencia de RPS19 se puede compensar con la sobreexpresión de RPS19 exógeno, explicándose así el fenómeno de la haploinsuficiencia y el aumento la posible terapia contra la enfermedad, aumentando el gen RPS” (1).
CLÍNICA
En los niños, esta enfermedad se presenta con signos derivados de la anemia: palidez muco-cutánea, retraso en el crecimiento, dificultades para la alimentación. También, “dependiendo del gen mutado, la anemia puede ser clasificada en varios subtipos y puede manifestarse de forma diferente” (5). Además de esto, en un 50% de los pacientes con esta enfermedad, se presentan malformaciones congénitas como: labio leporino, fisura palatina, pulgar trifalángico patognomónico, deformidades en los pabellones auriculares, malformaciones cefálicas por retraso en el cierre de las fontanelas, afectación del corazón y del tracto urogenital (hipospadias). Se ha testimoniado hipotrofia al nacer y retraso del crecimiento en el 28% y el 22%, respectivamente. La baja estatura presente durante la vida intrauterina en el 22% de los casos, empeora progresivamente debido a los efectos secundarios del tratamiento de la enfermedad (esteroides y sobrecarga de hierro por transfusiones).

Ilustración 2: Malformaciones congénitas en la anemia de Blackfan-Diamond. Tomado de : Hidalgo et al. "Importance interdisciplinary management of Diamond-Blackfan anemia and Cleft lip and palate sequel: Accompaniment of two years". Revista de Odontopediatría Latinoamericana (4). Ilustración 3: Tomado de : healthjade.net/diamond-blackfan-anemia (6).
DIAGNÓSTICO
Hemograma completo y reticulocitos
En el hemograma, vemos una anemia normocrómica y normalmente macrocítica. Esto sumado a la reticulocitopenia (<20.109/L) es una característica importante en estos pacientes. El recuento de neutrófilos y plaquetas suele ser normal, aunque se ha descrito trombocitosis transitoria en niños pequeños, así como trombocitopenia y leuconeutropenia leves en el 25% de los casos.
Hemoglobina fetal
Los niveles de hemoglobina fetal están elevados después de los 6 meses de edad y pueden guiar el diagnóstico.
Aspirado o biopsia de médula ósea.
La anemia de Blackfan-Diamond se diagnostica mediante la presencia de eritroblastopenia en aspirados o biopsias de médula ósea. La ausencia o <5% de precursores eritroides en el frotis o la disminución de celularidad eritroide en la biopsia, con el resto de celularidad normal (y sin signos de displasia) es diagnóstico.
eADA
La actividad de la adenosina desaminasa eritroide (eADA) es útil para tanto para el diagnóstico como para el asesoramiento genético y la elección de un donante familiar para el TPH. En los pacientes con esta enfermedad que aún no han sido transfundidos, la actividad se encuentra elevada en un 75%-90%. La elevación de eADA en un padre, orienta hacia una forma familiar de la enfermedad. No obstante, aún no se ha definido el vínculo entre este marcador y la Anemia de Blackfan-Diamond. “La actividad debe medirse en pacientes antes de la transfusión, y en caso de ya haberse hecho, se debe esperar al menos 3 meses después” (2).
Diagnóstico molecular
Tanto el genotipo como el fenotipo de la anemia de Blackfan-Diamond soy muy heterogéneos. La herencia es autosómica dominante en el 45% de los casos y esporádica o familiar con patrones de herencia diferentes en el porcentaje restante. Se han identificado herencias recesivas (genes EPO) o ligadas al cromosoma X (genes GATA1, TSR2).
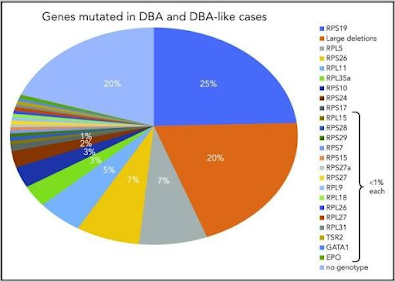
Ilustración 4: Genes mutados en la anemia de Blackfan-Diamond. Tomado de : Da Costa, Lydie et al. “Anemia de Diamond-Blackfan”. Blood 136,11 (2020): 1262-1273. doi:10.1182/blood.2019000947 (2).
La penetración de esta enfermedad es incompleta. Por lo tanto, la gravedad clínica es variable, va desde fenotipos silenciosos sin anemia hasta fenotipos de enfermedad leves y graves con anemia. Los fenotipos silenciosos son portadores de una mutación en un gen RP o bien muestran características biológicas de la enfermedad como macrocitosis aislada, aumento de la hemoglobina fetal o actividad elevada de eADA, pero sin presentar anemia. Sin embargo, este fenotipo tiene el mismo riesgo de transmisión que los portadores de la enfermedad anémica y, por lo tanto, deben identificarse para asesoramiento genético o antes de la selección como donantes de progenitores hematopoyéticos. Además, no están exentos de eventos hematológicos (aparición tardía de anemia o neoplasias malignas hematológicas) y tumores sólidos.
Genes implicados
El gen RPS19 fue el primero identificado en la anemia de Blackfan-Diamond en el año 1990 y estudios posteriores mostraron que se encuentra en un 25% de los pacientes afectados por esta enfermedad. A principios de la década del 2000 se identificaron mutaciones de un conjunto de genes ribosómicos que codifican constituyentes de las subunidades ribosómicas: RPL5 (7% de los casos), RPL11 (5%), RPL35a (3,5%), RPS24 (2,4%) … En total han sido identificados 64 genes, sin embargo, el gen RPS19 es el que con más frecuencia se ve afectado y todas las mutaciones identificadas son heterocigotas, pues en homocigosis se considera incompatible con la vida.
Existen genes identificados no RP asociados con un fenotipo similar a la anemia de Blackfan-Diamond, que incluyen el gen GATA1, TSR2, EPO y ADA2. El gen GATA1 es el principal factor de transcripción eritroide y la mutación relacionada con el fenotipo de
la enfermedad provoca una omisión del exón 2 y la pérdida del dominio de transactivación.
Criterios diagnósticos
Dentro de los criterios diagnósticos para la anemia de Blackfan-Diamond clásica se encuentran los siguientes puntos:
- Edad de inicio menor de 12 meses.
- Anemia macrocítica sin otras citopenias significativas.
- Reticulocitopenia.
- Médula ósea con celularidad normal con falta de precursores eritroides.
A parte de estos, hay otros criterios principales de apoyo:
- Mutación genética descrita para la anemia de Blackfan-Diamond.
- Antecedentes familiares de esta enfermedad.
Y, por último, encontramos criterios diagnósticos menores:
- Actividad elevada de eADA.
- Anomalías congénitas descritas en la anemia de Blackfan-Diamond clásicas.
- Hemoglobina fetal elevada.
- No evidencia de otro síndrome de insuficiencia medular hereditario.
DIAGNÓSTICO DIFERENCIAL
El diagnóstico diferencial de esta enfermedad debe hacerse sobre todo con otros síndromes de insuficiencia medular como los siguientes:
- Eritroblastopenia transitoria de la infancia: un tipo de anemia adquirida de causa desconocida que se presenta al año de edad en el 80% de los casos (a diferencia de la anemia de Blackfan-Diamond, con el 90% de pacientes diagnosticados antes del año de edad). Otra diferencia distintiva es que, en la eritroblastopenia transitoria, tan solo un 10% de los pacientes tienen eADA elevado y la anemia suele ser normocítica.
- Anemia de Fanconi: cursa con pancitopenia y las anomalías físicas congénitas se presentan a lo largo de la primera década de la vida.
- Síndrome de Shwachaman-Diamond: síndrome clínico caracterizado por disfunción pancreática exocrina con malabsorción intestinal, citopenia de una o varias estirpes celulares, retraso del crecimiento, anomalías óseas y susceptibilidad a síndromes mielodisplásicos y leucemia mieloide aguda.
- Síndrome de Pearson: mutación del mDNA de herencia materna que cursa con una anemia sideroblástica de la infancia, insuficiencia pancreática exocrina, insuficiencia hepática, defectos de los túbulos renales y pancitopenia.
- Disqueratosis congénita: triada clásica de pigmentación reticular en encaje en la parte superior del tórax, uñas displásicas y leucoplasia oral.
- Displasia del cartílago-cabello: trastorno hereditario autosómico recesivo que se caracteriza por anemia, macrocitosis, respuesta defectuosa de los linfocitos T, huesos tubulares cortos y pelo rubio muy fino y escaso.
- Síndrome de trombocitopenia con ausencia de radio: se presenta en el período neonatal con trombocitopenia, ausencia de radio y presencia de pulgares bilaterales. Otros hallazgos como deformidades en las costillas, cervicales, cubitales, humerales… En casos raros, cursa con anemia aplásica, citopenias o leucemia.
- Trombocitopenia amegacariocítica congénita: se presenta al nacer o en el período neonatal con trombocitopenia severa, petequias y sangrado intracraneal o de la mucosa intestinal. Durante la infancia pueden desarrollar pancitopenia, síndrome mielodisplásico o leucemia.
El parvovirus B19 es una causa común de insuficiencia de la médula ósea, que cursa con anemia grave en niños con hemólisis crónica, por lo que “es obligatorio realizar una serología de parvovirus B19 o PCR de parvovirus B19 en sangre en pacientes en los que se sospeche anemia de Blackfan-Diamond” (1).
TRATAMIENTO Y NUEVOS AVANCES
Estos pacientes necesitan un tratamiento multidisciplinario, que evolucione en función de la edad del paciente y específico para cada grupo de edad. Puesto que, a parte del tratamiento hematológico, se deben manejar las anomalías congénitas, el retraso en el crecimiento, los efectos adversos del tratamiento de la propia enfermedad (toxicidad de corticoides y sobrecarga férrica por transfusiones sanguíneas). De esta manera, también deben tratarse y prevenir las complicaciones a largo plazo, sobre todo en los pacientes adultos, como la inmunodeficiencia de células B y neoplasias malignas. Sin embargo, “algunas personas con enfermedad leve no necesitan tratamiento” (5).
Tratamiento de la anemia
Tras el nacimiento y en la infancia, los niveles de hemoglobina pueden ser normales o estar disminuidos, pero el 90% de los pacientes suele necesitar transfusiones antes de cumplir el año de edad. No está aprobada la utilización de corticoides antes del año de edad, incluso puede retrasarse a partir del año en niños con retrasos graves del crecimiento. El umbral a partir del cual se realizan las transfusiones es con una hemoglobina entre 80-90 g/L.
En niños que superan el año, los glucocorticoides pueden ser pautados a dosis estándar (2 mg/kg). En aquellos pacientes que responden a este tratamiento, los reticulocitos aumentan a partir del décimo día y el valor de hemoglobina vuelve a la normalidad 1 mes después. Tras ello, los corticoides deben reducirse gradualmente hasta definir la dosis mínima activa. Entre el 50% y el 60% responden a largo plazo a los corticoides. La anemia de Blackfan-Diamond es la única enfermedad humana en la que se administran glucocorticoides durante años, por lo que la eficacia y efectos adversos deben evaluarse periódicamente. Pues se consideran criterios suficientes para la suspensión del tratamiento los siguientes: retraso del crecimiento en preadolescentes, pues la retirada de los corticoides permitirá un aumento de la eficacia de la terapia con hormona del crecimiento, si estuvieses indicada; en adultos en quienes se observen efectos secundarios a largo plazo por los esteroides. La resistencia a estos fármacos ocurre en un 35% de los pacientes, mostrando una respuesta de reticulocitos mínima o incluso nula, sin cambios en los niveles de hemoglobina. Estos pacientes son candidatos para recibir transfusiones de concentrados de hematíes, así como candidatos para trasplante de células madre hematopoyéticas.
Para aquellos pacientes que reciben transfusiones sanguíneas, tienen un riesgo muy alto de sobrecarga férrica. Se ha llegado a observar hemocromatosis grave en pacientes jóvenes con anemia de Blackfan-Diamond. Por lo tanto, se debe pautar una terapia con quelantes de hierro en estos pacientes incluso a edades tempranas, normalmente antes de los 2 años, dependiendo esto sobretodo de la edad de la primera transfusión y los niveles de ferritina. La Deferoxamina es el fármaco quelante de elección, cuyo objetivo es lograr disminuir los niveles de ferritina a <500µg/L y normalizar el hierro en el hígado.
Actualmente, el trasplante de progenitores hematopoyéticos es la única terapia curativa para esta enfermedad y debe considerarse en fases tempranas en niños con dependencia de transfusiones en edades de 3 a 5 años, con mala respuesta a corticoides o toxicidad a los mismos.
La terapia génica es otra opción terapéutica potencial en la anemia de Blackfan- Diamond. Se centra en el gen RPS19, con éxito en modelos con ratones, pero actualmente sin ensayos activos en humanos.
Manejo de las complicaciones
Estos pacientes necesitan una atención clínica integral por parte de muchas especialidades médicas. Puesto que en los niños es fundamental el crecimiento, los esteroides limitan mucho el tratamiento. Se debe considerar pautar leucina y hormona del crecimiento a niños con retrasos muy importantes, siempre con vigilancia y precaución.
Sin embargo, son los adultos los que tienen un mayor riesgo de complicaciones graves, pues tienen mayor susceptibilidad a las neoplasias malignas. La incidencia acumulada de tumores sólidos y neoplasias malignas hematológicas en estos pacientes es de 13,7 a los 45 años. Esto respalda la necesidad de vigilancia específica del cáncer, como colonoscopias periódicas para cáncer de colon.
Los niveles de inmunoglobulinas deben controlarse anualmente, principalmente en pacientes adultos, puesto que al igual que otros síndromes de insuficiencia medular hereditarios, la anemia de Blackfan-Diamond puede cursar con inmunodeficiencia.
Últimos avances
En un estudio publicado en julio de 2020 (3) se identificó que la actividad quinasa similar a Nemo (NLK) se encuentra aumentada en los progenitores eritroides de los modelos ratón de anemia de Blackfan-Diamond. Y que la supresión de la actividad de esta quinasa puede dar lugar a la expansión de la serie eritroide. Por tanto, se muestra que la NLK es una parte fundamental de la patogenia de estar enfermedad, así como una diana importante a la hora de buscar tratamientos efectivos contra ella. Sin embargo, los fármacos probados, al inhibir de forma sistémica la NLK, presente también en tejidos cerebrales, causan severos efectos secundarios a nivel pulmonar y neurológico, en algunos casos llegando a observarse un desarrollo estromal aberrante en la médula ósea de ratones con total inhibición de NLK.
Concluimos por tanto que “la activación sostenida de NLK en modelos humanos y murinos con anemia de Blackfan-Diamond con mutaciones en diferentes subunidades ribosómicas y la inhibición química de NLK mejoró la eritropoyesis en todos los modelos examinados” (3).
PRONÓSTICO
El pronóstico es generalmente bueno. Sin embargo, las complicaciones del tratamiento y una elevada incidencia de tumores pueden recudir la esperanza de vida, así como alterar la morbilidad. La gravedad de la enfermedad, por tanto, depende de la calidad de los cuidados recibidos y la respuesta al tratamiento. Para pacientes que reciben transfusiones regulares, la calidad de vida se ve alterada.
BIBLIOGRAFÍA
1. Gadhiya K, Wills C. Diamond Blackfan Anemia. 2022 Jan 31. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan–. PMID: 31424886.
2. Da Costa L, Leblanc T, Mohandas N. Diamond-Blackfan anemia. Blood. 2020 Sep 10;136(11):1262-1273. doi: 10.1182/blood.2019000947. PMID: 32702755; PMCID: PMC7483438.
3. Wilkes, M. C., Siva, K., Chen, J., Varetti, G., Youn, M. Y., Chae, H., Ek, F., Olsson, R., Lundbäck, T., Dever, D. P., Nishimura, T., Narla, A., Glader, B., Nakauchi, H., Porteus, M. H., Repellin, C. E., Gazda, H. T., Lin, S., Serrano, M., Flygare, J., … Sakamoto, K. M. (2020). Diamond Blackfan anemia is mediated by hyperactive Nemo-like kinase. Nature communications, 11(1), 3344.
4. Hidalgo, C. A. C., Guaman, G. S., & Pardo, M. D. P. B. (2020). Importance interdisciplinary management of Diamond-Blackfan anemia and Cleft lip and palate sequel: Accompaniment of two years. Revista de Odontopediatría Latinoamericana, 9(1), 66-74.
5. Anemia de Diamond-Blackfan | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program.
6. Diamond-Blackfan Anemia - Causes, Symptoms, Diagnosis & Treatment – Health Jade.
No hay comentarios:
Publicar un comentario
Se admiten comentarios que indiquen posibles errores en los textos y/o sugerencias de temas, y/o propuestas de mejoras en el blog y/o dudas sobre la realización de trabajos. Dado que este blog no es un consultorio médico, no se responderán preguntas realizadas con esa finalidad. Los comentarios que se consideren inapropiados, serán eliminados de inmediato, sin ningún tipo de excepción.
Nota: solo los miembros de este blog pueden publicar comentarios.