Autora : Isabel Ortiz Carrascosa
4º Curso Medicina grupo "A" (curso 2021-2022)
Código de trabajo : 2102-IOC
Introducción
La anemia de Blackfan-Diamond es una enfermedad hereditaria hematológica incluida dentro del conjunto de enfermedades raras, caracterizada por una aplasia medular arregenerativa congénita con eritroblastopenia que se manifiesta en los primeros meses de vida, pudiendo ser diagnosticada hasta los 2 años de vida y muy rara vez después de los 4 años (1).
Por ello, los pacientes tienen un déficit de la serie roja que produce severas consecuencias, además de otras malformaciones.
Por ello, los pacientes tienen un déficit de la serie roja que produce severas consecuencias, además de otras malformaciones.
Aplasias medulares
Para entender esta enfermedad, hay que entender el concepto de aplasia o insuficiencia medular, en la que hay un déficit de una o más series de células sanguíneas por diversas causas a nivel central en la médula ósea. En el caso de la anemia de Blackfan-Diamond, se trata de una insuficiencia medular selectiva, pues solo afecta a la serie roja (anemia).
La aplasia pura de la serie roja o eritroblastopenia, se caracteriza por:
- Anemia normocítica severa.
- Reticulocitos < 20.000/mm3.
- Ausencia selectiva de precursores eritroides en la médula ósea.
- La celularidad medular es normal, con cifras de leucocitos y plaquetas normales.
A su vez, las eritroblastopenias pueden ser:
- Congénitas: anemia de Blackfan-Diamond (tema a tratar).
- Adquiridas: bien idiopáticas o secundarios a otras causas (fármacos, infecciones, etc).
Clínica
En general, los pacientes suelen ser diagnosticados antes de los 18 meses de vida. Los primeros síntomas de alarma suelen ser palidez y disnea durante la alimentación o lactancia. Dicha palidez es aislada, no se encuentran visceromegalias ni otros signos de hemolisis.
También aparece una anemia macrocítica arregenerativa severa, siendo las demás series hematopoyéticas normales (2). Los pacientes suelen presentar una baja estatura y anomalías congénitas, sobre todo a nivel craneofacial (secuencia de Pierre-Robin y fisura palatina), urogenital (ausencia de riñón o riñón en herradura), cardiológico (4), ocular (cataratas, glaucoma, esclerótica azul), en los pulgares (frecuente que tengan tres falanges) (1). Estas malformaciones aumentan el riesgo de padecer otras enfermedades neoplásicas en el futuro, bien por tumores sólidos o hematológicos; además de problemas hormonales tales como insuficiencia suprarrenal, hipogonadismo e hipotiroidismo (2).
También aparece una anemia macrocítica arregenerativa severa, siendo las demás series hematopoyéticas normales (2). Los pacientes suelen presentar una baja estatura y anomalías congénitas, sobre todo a nivel craneofacial (secuencia de Pierre-Robin y fisura palatina), urogenital (ausencia de riñón o riñón en herradura), cardiológico (4), ocular (cataratas, glaucoma, esclerótica azul), en los pulgares (frecuente que tengan tres falanges) (1). Estas malformaciones aumentan el riesgo de padecer otras enfermedades neoplásicas en el futuro, bien por tumores sólidos o hematológicos; además de problemas hormonales tales como insuficiencia suprarrenal, hipogonadismo e hipotiroidismo (2).

Figura 1. Secuencia de Pierre-Robin: micrognatia, glosoptosis y
obstrucción respiratoria alta con o sin paladar hendido.

Figura 2. Pulgar con tres falanges, característico.
Etiología
Es una enfermedad que se hereda de forma autosómica dominante, por lo que solo hace falta una copia anormal del gen mutado para que se manifieste la enfermedad. A pesar de esto, también puede aparecer ‘de novo’ en una familia debido a una mutación puntual.
En los casos hereditarios, se han encontrado genes que están presentes en un 45% de los pacientes. Todos ellos codifican proteínas ribosómicas o de la subunidad pequeña (RPS7, RPS17, RPS19 en un 25% de los casos, RPS24) o grande (RPL5, RPL11, RPL35A) del ribosoma (1).
En casos muy raros, puede estar causado por una mutación en el gen GATA1, gen que codifica un factor de transcripción involucrado en el desarrollo eritroide (4), o deberse a una herencia ligada al cromosoma X (2).
Dependiendo de cuál sea la mutación implicada, esta enfermedad puede clasificarse en diferentes subtipos.
En los casos hereditarios, se han encontrado genes que están presentes en un 45% de los pacientes. Todos ellos codifican proteínas ribosómicas o de la subunidad pequeña (RPS7, RPS17, RPS19 en un 25% de los casos, RPS24) o grande (RPL5, RPL11, RPL35A) del ribosoma (1).
En casos muy raros, puede estar causado por una mutación en el gen GATA1, gen que codifica un factor de transcripción involucrado en el desarrollo eritroide (4), o deberse a una herencia ligada al cromosoma X (2).
Dependiendo de cuál sea la mutación implicada, esta enfermedad puede clasificarse en diferentes subtipos.

Tabla 1. Subtipos de la Anemia de Blackfan-Diamond según la mutación genética implicada (3).
A pesar de que la mayoría de estas mutaciones solo se encuentran en un porcentaje muy bajo de pacientes, existe una correlación entre la aparición de anomalías craneoencefálicas en portadores de RPL5 y RPL11 y la no aparición de estas anomalías en portadores de RPS19 (1).
Patogenia
La enfermedad se produce por un trastorno intrínseco de la eritropoyesis, con una disminución de las CFU-E y en algunos casos, también de las BFU-E debido a una hipersensibilidad a la muerte por apoptosis (8) por la falta de funcionamiento de las proteínas ribosomales y/o errores en su ensamblaje. Una explicación viene dada por la ‘Hipótesis del estrés ribosómico’. En condiciones normales, una proteína llamada HDM2 (ubiquitina ligasa) regula los niveles y actividad del p53, promoviendo su degradación. La interacción de esta proteína junto con otras proteínas mutadas que aparecen en la Anemia de Blackfan- Diamond (RPL5, RPL11 y RPL23) reducirían su actividad e impedirían la degradación del p53, afectando así a los precursores eritroides.
Criterios diagnósticos (6)
1. Edad menor de 1 año.
2. Anemia macrocítica sin otras citopenias.
3. Reticulocitopenia.
4. Celularidad normal en médula ósea, con escasez de precursores eritroides (5)
2. Anemia macrocítica sin otras citopenias.
3. Reticulocitopenia.
4. Celularidad normal en médula ósea, con escasez de precursores eritroides (5)

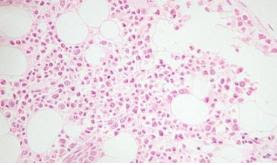
Criterios de soporte:
- Historia familiar: se puede confirmar hasta en un 20% de los casos (1).
- Detección de las mutaciones causantes (gran valor diagnóstico).
- Aumento de actividad de adenosina deaminasa (ADA) eritrocitaria: es frecuente pero no es específica, puede elevarse en personas que no padezcan la enfermedad.
- Anomalías congénitas características con la enfermedad.
- Hemoglobina F elevada.
- No evidencia de otros fallos medulares congénitos.
- Historia familiar: se puede confirmar hasta en un 20% de los casos (1).
- Detección de las mutaciones causantes (gran valor diagnóstico).
- Aumento de actividad de adenosina deaminasa (ADA) eritrocitaria: es frecuente pero no es específica, puede elevarse en personas que no padezcan la enfermedad.
- Anomalías congénitas características con la enfermedad.
- Hemoglobina F elevada.
- No evidencia de otros fallos medulares congénitos.
Diagnóstico diferencial
- Eritroblastopenia transitoria de la infancia: se da en niños mayores de un año, no es de carácter hereditario y la HbF y la ADA no están elevados (8).
- Otras ribosomopatías: disqueratosis congénita, hipoplasia del cartílago- cabello, síndrome de Shwachman-Diamond, síndrome de Treacher Collins, síndrome de Bowen-Conradi, cirrosis infantil de los indios de Norte América (6).
- Otras ribosomopatías: disqueratosis congénita, hipoplasia del cartílago- cabello, síndrome de Shwachman-Diamond, síndrome de Treacher Collins, síndrome de Bowen-Conradi, cirrosis infantil de los indios de Norte América (6).
Tratamiento
- Corticoides (Prednisona): mejora la anemia en el 80% de los casos, en los casos donde no haya mejora, deben suspenderse poco a poco (6). Contraindicados en el primer año de vida (1).
- Transfusiones de sangre: para mejorar la anemia y mantener una concentración de hemoglobina y glóbulos rojos óptima, además de mantener el crecimiento y el desarrollo del niño.
- Trasplante de médula ósea con HLA idéntico de hermano no afectado (1): único tratamiento potencialmente curativo. A pesar de esto, no desaparece el riesgo aumentado de padecer otras neoplasias (las malformaciones ya están presentes).
- Transfusiones de sangre: para mejorar la anemia y mantener una concentración de hemoglobina y glóbulos rojos óptima, además de mantener el crecimiento y el desarrollo del niño.
- Trasplante de médula ósea con HLA idéntico de hermano no afectado (1): único tratamiento potencialmente curativo. A pesar de esto, no desaparece el riesgo aumentado de padecer otras neoplasias (las malformaciones ya están presentes).
Pronóstico
El pronóstico es generalmente bueno (1), aunque depende fundamentalmente de la clínica del paciente, la gravedad de las alteraciones morfológicas, la severidad de la anemia, la respuesta al tratamiento y las complicaciones asociadas. A largo plazo, hay mayor riesgo de padecer diferentes neoplasias, posiblemente por las ventajas de los clones con mutaciones en p53.
Las complicaciones asociadas al tratamiento suelen ser por efectos secundarios del uso prolongado de corticoides, sobrecarga de hierro por las transfusiones, etc.
Debido a los avances, ha aumentado el número de mujeres embarazadas con esta patología. Estos embarazos son considerados de alto riesgo por retraso en el crecimiento fetal intrauterino, frecuencia de pérdidas fetales, prematuridad, preeclampsia y otros; por ello deben ser monitoreados y manejados de forma multidisciplinar por obstetras, hematólogos y neonatólogos (9).
Las complicaciones asociadas al tratamiento suelen ser por efectos secundarios del uso prolongado de corticoides, sobrecarga de hierro por las transfusiones, etc.
Debido a los avances, ha aumentado el número de mujeres embarazadas con esta patología. Estos embarazos son considerados de alto riesgo por retraso en el crecimiento fetal intrauterino, frecuencia de pérdidas fetales, prematuridad, preeclampsia y otros; por ello deben ser monitoreados y manejados de forma multidisciplinar por obstetras, hematólogos y neonatólogos (9).
Bibliografía
1. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=ES&Expert=124
2. https://rarediseases.info.nih.gov/espanol/12933/anemia-de-diamond- blackfan
3. https://omim.org/phenotypicSeries/PS105650
4. Ellis, S. R. (2014). Nucleolar stress in diamond blackfan anemia pathophysiology. Biochimica et Biophysica Acta - Molecular Basis of Disease, 1842(6), 765–768. https://doi.org/10.1016/j.bbadis.2013.12.013
5. https://www.medigraphic.com/pdfs/pinar/rcm-2017/rcm174p.pdf
6. Machado, A. (2010). Muchas gracias a todos. 2010.
7. Presentación Hematología 2021. Medicina.
8. Lipton, J. M., & Ellis, S. R. (2009). Diamond-Blackfan Anemia: Diagnosis, Treatment, and Molecular Pathogenesis. Hematology/Oncology Clinics of North America, 23(2), 261–282. https://doi.org/10.1016/j.hoc.2009.01.004 Rich. (2009). NIH Public Access. Bone, 23(1), 1–7. https://doi.org/10.1016/j.tracli.2010.06.001.Diamond-Blackfan
2. https://rarediseases.info.nih.gov/espanol/12933/anemia-de-diamond- blackfan
3. https://omim.org/phenotypicSeries/PS105650
4. Ellis, S. R. (2014). Nucleolar stress in diamond blackfan anemia pathophysiology. Biochimica et Biophysica Acta - Molecular Basis of Disease, 1842(6), 765–768. https://doi.org/10.1016/j.bbadis.2013.12.013
5. https://www.medigraphic.com/pdfs/pinar/rcm-2017/rcm174p.pdf
6. Machado, A. (2010). Muchas gracias a todos. 2010.
7. Presentación Hematología 2021. Medicina.
8. Lipton, J. M., & Ellis, S. R. (2009). Diamond-Blackfan Anemia: Diagnosis, Treatment, and Molecular Pathogenesis. Hematology/Oncology Clinics of North America, 23(2), 261–282. https://doi.org/10.1016/j.hoc.2009.01.004 Rich. (2009). NIH Public Access. Bone, 23(1), 1–7. https://doi.org/10.1016/j.tracli.2010.06.001.Diamond-Blackfan
No hay comentarios:
Publicar un comentario
Se admiten comentarios que indiquen posibles errores en los textos y/o sugerencias de temas, y/o propuestas de mejoras en el blog y/o dudas sobre la realización de trabajos. Dado que este blog no es un consultorio médico, no se responderán preguntas realizadas con esa finalidad. Los comentarios que se consideren inapropiados, serán eliminados de inmediato, sin ningún tipo de excepción.
Nota: solo los miembros de este blog pueden publicar comentarios.