4º Curso Medicina grupo A (Curso 2021-2022)
Código de trabajo : 2104-CPS

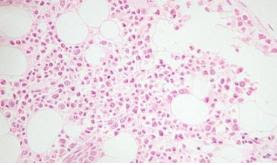
Figura 1: Frotis de sangre periférica con presencia de drepanocitos (imagen obtenida del Atlas del Grupo Español de Citología Hematológica)
Código de trabajo : 2104-CPS
Introducción
La hemoglobina (Hb) es una proteína formada por cuatro cadenas peptídicas cada una de ellas con un grupo hemo como grupo prostético. El grupo hemo es una protoporfirina con un átomo de Fe en su centro.
Existen distintas combinaciones de cadenas peptídicas capaces de formar hemoglobina siendo las más importantes:
· HbA: formada por dos cadenas alfa y dos betas (α2β2), y que supone el 97% de la Hb total en el adulto
· HbA2: formada por dos cadenas alfa y dos delta (αδ), y que supone aproximadamente un 2,5% de la Hb total en el adulto.
· HbF: también llamada hemoglobina fetal es la mayoritaria en la hematopoyesis intrauterina, está formada por dos cadenas alfa y dos cadenas gamma (αγ), y en adulto supone menos del 1% de la hemoglobina total.
Cada una de las cadenas de hemoglobina se sintetiza en su propio gen. La cadena alfa se sintetiza en el brazo corto del cromosoma 16, conteniendo dos copias en cada cromosoma, mientras que los genes de las cadenas beta, gamma y delta se localizan en el brazo corto del cromosoma 11, existiendo una única copia de cada uno por cromosoma. (1)
Otro concepto previo a tener en cuenta sería el de hemoglobinopatía, que de acuerdo con el Manual de Hematología Clínica (2) se define como defecto hereditario de la Hb por mutaciones en los genes de las globinas. Existen varios tipos según se deban a un déficit cuantitativo de la síntesis de una de las cadenas o estructural por cambio en la secuencia de aminoácidos.
Etiopatogenia
La drepanocitosis es una hemoglobinopatía estructural que se produce por una mutación puntual consistente en el cambio de una base nitrogenada (timina en lugar de adenina) que implica un cambio en el sexto aminoácido de la cadena β de hemoglobina, cambiando un ácido glutámico por una valina). Este cambio de aminoácido repercute en que se pierde una carga negativa (presente en el ácido glutámico) lo que altera la conformación tridimensional de la cadena β.
La hemoglobina resultado de esta mutación recibe el nombre de Hemoglobina S (Hb S), y se caracteriza porque en estados con baja presión de oxígeno tiene la capacidad de polimerizar formando filamentos de hemoglobina que deforman el eritrocito confiriéndole la “forma de hoz” característica de los hematíes de la enfermedad. Dichos hematíes deformados reciben el nombre de drepanocitos (fig 1). Otros factores como el frío, bajos niveles de pH, deshidratación e infecciones también pueden favorecer la precipitación de la HbS. (3)(4)
Figura 1: Frotis de sangre periférica con presencia de drepanocitos (imagen obtenida del Atlas del Grupo Español de Citología Hematológica)
La mutación se transmite de forma autosómica dominante (como en todas las hemoglobinopatías en general), pero en el caso de los heterocigotos únicamente heredan el rasgo drepanocítico, de tal forma que en general son portadores asintomáticos que únicamente presentarán cierto grado anemia leve y solo desarrollarían síntomas en situaciones de anoxia. Los pacientes afectados por tanto, serán los homocigotos (o los dobles heterocigotos, en caso de que el gen no afecto por la mutación anteriormente descrita sea anormal por otra hemoglobinopatía) los cuales desarrollarán la anemia drepanocítica como tal.
Epidemiología
Merece la pena comentar la distribución epidemiológica de la anemia de células falciformes, cuya prevalencia es mayor en el África Subsahariana, donde la mutación genética está presente en aproximadamente un 40% de la población. También existe un porcentaje significativo en algunos núcleos Mediterráneos como Grecia, Turquía, Italia y Norte de África. En América es más frecuente encontrar la mutación en descendientes de emigrantes africanos (población afro-americana) dada la mayor prevalencia que existente en ente grupo poblacional. Actualmente la India también es un foco epidemiológico a tener en cuenta.
El mapa de distribución coincide con el de la incidencia de malaria, ya que al igual que ocurre con otras hemoglobinopatías como las talasemias, el ser heterocigoto para la drepanocitosis constituye un factor protector frente a la infección por Plasmodium falciparum. (3)(5)
Fisiopatología
La deformación del hematíe le hace perder su forma redonda y bicóncava, esto provoca que tengan mayor capacidad de adherencia y que pierdan su capacidad de deformarse. También es típico el aumento en la viscosidad de la sangre por interacciones anómalas entre los eritrocitos y el resto de componentes sanguíneos así como el endotelio vascular. Este aumento de adherencia se cree que es provocado por una pérdida de la asimetría de la membrana del eritrocito, aumentándose la exposición de fosfatidilserina en la superficie. (5)
Todo ello contribuye a favorecer la agrupación de los drepanocitos entre sí y su adherencia al endotelio vascular provocando vaso-oclusión (fig 2). Es por ello que la principal clínica derivada de la enfermedad será resultado de crisis vasooclusivas, con el consiguiente daño orgánico que provoque.

Figura 2: Explicación de las crisis vasooclusivas de células falciformes (Creason C. Stedman´s Medical Terminology. 2nd ed. Baltimore. Wolters Kluwer Health. 2011
Las complicaciones más frecuentes son las siguientes:
Algunos autores también sugieren que también existe una predisposición a la hemólisis intravascular traumática, con liberación de Hb al torrente sanguíneo, que se podría relacionar con una inhibición del NO y con ello un descenso de la vasodilatación, aumentando con ello el riesgo de complicaciones.
Otro aspecto relevante en la fisiopatología es el papel de la proteína banda 3 eritrocitaria. La banda 3 es una familia de intercambiadores aniónicos presentes en la membrana de todas las células. En el eritrocito estas proteínas se agregan en la superficie del eritrocito como resultado de la oxidación desnaturalización de la Hb eritrocitaria, cuando esto sucede se genera un oligómero de esta proteína que es reconocido por anticuerpos naturales de tipo IgM que mediante la acción del complemento permiten la hemolisis extravascular por macrófagos esplénicos. Este es el proceso fisiológico que sufren los eritrocitos senescentes, pero también se ha documentado una mayor actividad de este mecanismo en pacientes de varias hemoglobinopatías, y más concretamente de la drepanocitosis. Este fenómeno estaría involucrado en dos procesos claves de la fisiopatología de esta enfermedad como son la hemólisis temprana de los drepanocitos y las crisis vasooclusivas. Sobre todo es relevante el segundo supuesto y es que varios estudios (5)(6) relacionan un descenso de los niveles séricos de anticuerpos (Ac) contra la proteínas de banda 3 eritrocitaria por consumo de los mismos. Las hipótesis actuales sugieren que la adhesión de estos Ac podrían ser un mecanismo que favorezca la no adhesión de los drepanocitos y por tanto parte de la solución a las crisis oclusivas típicas de la enfermedad. Asimismo también se abriría una ventana al posible desarrollo de estrategias terapéuticas basadas en este mecanismo. (6)
Clínica
Al nacimiento los pacientes son asintomáticos por la persistencia de la Hb fetal, apareciendo las primeras manifestaciones de la enfermedad entre los 4 y 6 meses de vida. El cuadro clínico viene determinado por una hemólisis crónica con agudización en procesos con aumento del estrés oxidativo, y por el aumento de la adhesión eritrocitaria, que puede producir la oclusión de vasos dando una clínica muy variada según el órgano afecto, y siendo la principal causa de las complicaciones de la enfermedad.
Las complicaciones más frecuentes son las siguientes:
· Crisis vaso-oclusivas:
Son fenómenos de oclusión circulatoria desencadenados en su mayoría por la presencia de factores que aumenten la proporción de drepanocitos en sangre (situaciones que produzcan un aumento del estrés oxidativo) y con ello la probabilidad de adhesión eritrocitaria y posterior isquemia circulatoria. Dichas crisis pueden afectar a cualquier vaso sanguíneo, produciendo diferente clínica según el órgano afecto. Lo más común son las crisis de dolor vaso-oclusivas óseas que cursan con dolor intenso por infartos óseos, y cuyo manejo normalmente requiere de un equipo multidisciplinar y analgesia con opioides por su intensidad. No es despreciable tampoco el porcentaje de pacientes que sufren episodios isquémicos cerebrales por este motivo, llegando al 11% de prevalencia en homocigotos menores de 11 años. (3)(8)
· Fallo esplénico:
Se produce una esplenomegalia desde edades tempranas cuya evolución natural es un proceso denominado “auto-esplenectomía” en el que el bazo va sufriendo fibrosis progresiva. Existe una variante de evolución aguda del fallo esplénico llamada secuestro esplénico agudo que se caracteriza por una esplenomegalia aguda con bicitopenia y colapso circulatorio por secuestro esplénico. El tratamiento se basa en la realización de transfusiones y el uso de expansores de volumen. Si este suceso es de repetición la esplenectomía es de elección. (3)
· Infecciones de repetición:
Son la principal causa de morbi-mortalidad en estos pacientes, y generalmente se debe al fallo esplénico que suelen presentar. Otro factor predisponente es la posible presencia de zonas isquémicas intestinal por vaso-oclusión, estableciendo “puertas de entrada” para organismos colonizadores.
· Síndrome del tórax (síndrome torácico) agudo:
Se define como la presencia de un infiltrado alveolar no atelectásico que comprometa, al menos, un segmento del pulmón junto con presencia de fiebre mayor de 38,5 ºC, taquipnea, tos o sibilancias. Se estima que el 50% de los homocigotos tendrán al menos un episodio a lo largo de su vida. La propensión a este síndrome en pacientes de drepanocitosis es explicado por una triada de procesos fisiopatológicos que se retroalimentan entre sí. En primer lugar tendría su papel la mayor propensión a sufrir infecciones por parte de estos pacientes, y que es también aplicable a las infecciones de origen respiratorio. La presencia de infecciones localizadas en el pulmón provoca a su vez una zona hipoventilada que favorece la polimerización de Hb y con ello la generación de drepanocitos con tendencia a la adhesión y posiblemente de crisis vaso-oclusivas. Dichos fenómenos vaso-oclusivos se podrían producir en la zona infartada aumentando la hipoperfusión de la misma. A su vez existe una teoría según la cual afectarían también a la microcirculación ósea pudiéndose liberar émbolos que viajen por el sistema venoso hasta la circulación pulmonar embolizando nuevo territorio pulmonar. Esta complicación supone la segunda causa de ingreso más habitual. El tratamiento de elección en estos pacientes es antibioterapia de amplio espectro asociado a un macrólido. (8)(9)(10)
Tratamiento
En la actualidad el único tratamiento curativo de la enfermedad es el trasplante alogénico de progenitores hematopoyéticos con un donante HLA idéntico, el cual queda reducido como tratamiento de elección en jóvenes de entre 12-16 años con drepanocitosis severa. Esta intervención ha demostrado una supervivencia entre el 85-90% según el estudio seleccionado. A pesar de los buenos datos en ese sentido, hay que destacar la dificultad para obtener un donante haploidéntico, ya que solo está disponible en aproximadamente un 20% de los casos (3). Es por ello que se ha llegado a probar el uso de progenitores del cordón umbilical (se considera que sus células son menos inmunorreactivas), y a pesar de que los resultados eran algo mejores en los pacientes trasplantados con progenitores hematopoyéticos de médula ósea de donante haploidéntico, los resultados fueron bastante buenos, con una supervivencia a dos años de todos los participantes en el estudio (44 pacientes de talasemia y anemia drepanocítica). (11)(12)
La terapia con trasfusión de hematíes también están indicadas en estos pacientes, y se pueden realizar de forma aguda o crónica. Las trasfusiones agudas quedan restringidas a crisis de la enfermedad como el secuestro esplénico mencionado anteriormente, o el síndrome del tórax agudo. Las trasfusiones crónicas en cambio se recomiendan en todos los pacientes homocigotos a fin de mantener los niveles de HbS por debajo del 30% y con ello disminuir el riesgo de crisis oclusivas. Se recomienda que los pacientes que reciban trasfusiones crónicas reciban a su vez tratamiento con quelantes del hierro a fin de evitar hemosiderosis.
También hay fármacos que han demostrado que pueden ser de utilidad para esta patología entre los que destacan los siguientes:
· Hidroxiurea: se ha visto que este fármaco citotóxico puede usarse para mantener los niveles de HbF más altos de lo habitual en adultos, algo que se relaciona con buen pronóstico en esta enfermedad ya que asocia menor número de complicaciones. Inhibe la síntesis de ADN, induciendo supresión medular lo que parece ser que favorece la producción de precursores que contienen HbF. Este tratamiento se debe hacer bajo un seguimiento estrecho por parte de un hematólogo.
· Ácido fólico: se utiliza sobre todo en el contexto de acentuación de la anemia hemolítica con eritropoyesis elevada, que se da en complicaciones como infecciones o en las crisis vaso-oclusivas.
Asimismo es relevante mencionar el importante papel que tiene la prevención en esta enfermedad, puesto que el mejor tratamiento posible de las complicaciones es que estas no se produzcan. En primer lugar han demostrado su eficacia los programas de tamizaje neonatal por cromatografía de alta resolución, que ayudan a un diagnóstico precoz y que redundan en un descenso de la morbimortalidad en comparación con los pacientes diagnosticados en etapas posteriores. Esto se debe de acompañar de un programa de educación a los padres para poder realizar una intervención clínica temprana en caso de complicaciones.
También está indicada la profilaxis antibiótica con penicilina oral desde los 3 meses hasta los 5 años para evitar las infecciones, disminuyendo la incidencia de varias patologías habituales desde su implantación en EEUU. Para evitar las infecciones otra medida clave a realizar es la vacunación temprana, especialmente la referente a patógenos encapsulados contra los que podrían tener un mayor riesgo de complicaciones respecto a la población general. (3)
Bibliografía
1. Peñuela OA. Hemoglobina: una molécula modelo para el investigador. Colombia Medica. 2005; 36. Available from: https://www.redalyc.org/pdf/283/28336313.pdf
2. Sanz MA, Carreras E. Manual práctico de hematología clínica. 6a. Antares; 2019.
3. Quintero M, Hernández AJ. Anemia de células falciformes. Revista Gastrohnup. 2012; 14 (2); S27-S35. Available from: https://bibliotecadigital.univalle.edu.co/handle/10893/5929
4. Rees D, Williams T, Lancet MG-T, 2010. Sickle-cell disease. Lancet. 2010; Available from: https://www.sciencedirect.com/science/article/pii/S014067361061029X?c asa_token=FD0pgUnnVucAAAAA:tbJuT7eZz15BvbIZO2DRdoBxNINNMrKnttkq SlS3OJfta9fFrqkG7D7IMB2PCGnX1JflIss-
5. Vacca VM, Blank L. Drepanocitosis: situación actual y perspectivas. Nursing . 2017 Nov 1; 34 (6): 32–9. Available from: https://www.elsevier.es/es- revista-nursing-20-articulo-drepanocitosis-situacion-actual-perspectivas- S0212538217301814
6. Arce Hernández AA, Villaescusa Blanco R, Merlín Linares JC, Guerreiro Hernández AM. Estudio seriado de anticuerpos naturales antibanda 3 en enfermos con drepanocitosis. Revista Cubana hematología, inmunología y hemoterapia. 2016; 32. Available from: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864- 02892016000300008
7. Villaescusa Blanco R, Arce Hernández AA, Merlín Linares JC, Herrera Rolo T, Espinosa Martínez E. Anticuerpos naturales anti banda 3: Participan en el fenómeno de vasooclusión de la drepanocitosis. Revista Cubana hematología inmunología y hemoterapia. 2007; Available from: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864- 02892007000100005
8. González Sánchez M, Mayordomo Colunga J, Larrea Tamayo E, González Muñiz S, Sariego Jamardo A. Crisis vaso-oclusivas, una complicación frecuente de la drepanocitosis. Boletín de pediatría. 2010; 50. Available from: https://www.sccalp.org/documents/0000/1676/BolPediatr2010_50_281- 284.pdf
9. Gladwin MT, Vichinsky E. Pulmonary Complications of Sickle Cell Disease. New England Journal of Medicine. 2008 Nov 20; 359 (21): 2254–65. Available from: http://www.nejm.org/doi/abs/10.1056/NEJMra0804411
10. Miller AC, Gladwin MT. Pulmonary Complications of Sickle Cell Disease. American Journal of Respiratory Critical Care Medicine. 2012 Jun 1; 185 (11): 1154. Available from: /pmc/articles/PMC3373067/
11. Locatelli F, Rocha V, Reed W, Bernaudin F, Ertem M, Grafakos S, et al. Related umbilical cord blood transplantation in patients with thalassemia and sickle cell disease. Blood. 2003 Mar 15; 101 (6): 2137–43. Available from: https://pubmed.ncbi.nlm.nih.gov/12424197/
12. Oringanje C, Nemecek E, Oniyangi O. Hematopoietic stem cell transplantation for people with sickle cell disease. Cochrane Database Syst Rev. 2020 Jul 3; 2020 (7).