Autora : Agatha Mora Pascual
4º Curso Hematología grupo "D" (Curso 2022/23)
Código de trabajo : 2218-AMP
‘Qui bene interrogat, bene diagnoscit;
qui bene diagnoscit, bene curat’
Leube 1898
INTRODUCCIÓN
¿Y si pudiéramos detener el tiempo?¿Y si pudiéramos revertirlo?
Bien es sabido que en el mundo existen cantidad de seres vivos mucho más longevos que el ser humano, quizás vox populi sean las tortugas que pueden vivir más allá de 150 años, pero esto no es más que la punta del iceberg, pues las ballenas de Groenlandia viven más de 200 años, el Mejillón de agua dulce más de 250 años, el Tiburón de Groenlandia más de 272 años, la Almeja Quahog del océano más de 500 años, el Coral negro más de 4.000 años, la Esponja de vidrio más de 10.000 años. Contemplar estas cifras como potenciales años de vida desde el punto de vista de un ser humano se hace, cuanto poco, difícil y complejo. Por ejemplo, si pensamos que estamos en el año 2023 d.C. apenas estaríamos a media vida de un Coral negro. Grecia, Roma, la Edad Media, la Revolución industrial, apenas serían un fragmento en nuestras vidas.
Pero esto no es todo, sino que, a pesar de lo sorprendente que pueda parecer la existencia de un ser vivo de más de 10.000 años, todavía podemos hallar algo aún más impactante que pudiera considerarse fantasía, pero no, actualmente se han reconocido dos especies potencialmente inmortales, la medusa Turritopsis Dohrnii que cuenta con la mayor parte de la fama y la Hidra, otro tipo de invertebrado que también se contempla como inmortal.
En el caso de la medusa esta es capaz de reproducirse, o si es dañada, devolver sus células a un estado madurativo anterior y volver a envejecer de forma indefinida (1,2).

Figura 1. Ciclo vital Turritopsis dohrnii
La Hidra, por otro lado, no es capaz de revertir el estado de sus células, pero está compuesta en una amplia parte por células madre, las cuales se regeneran continuamente, ya sea por duplicación o clonación, dando lugar así a su inmortalidad (2).

Figura 2. Hidra
Todo esto que parece sorprendente para muchos, es realmente un enorme abanico de posibilidades y de oportunidades en el campo de la investigación, actualmente en desarrollo, de hecho, a finales de 2022 se logró secuenciar por primera vez el genoma de la medusa, por lo que nos podríamos plantear muchas cuestiones ¿Y si pudiéramos manipular nuestras células madre de la misma forma que lo hace la Hidra? ¿Y si pudiéramos rejuvenecer como lo hace la medusa?¿Y si digo que esto último ya pudiera ser una realidad?
CÉLULAS MADRE PLURIPOTENTES INDUCIDAS (iPS)
Estas ideas, que puedan resultar a primera vista únicamente fruto de la fantasía o de la ignorancia, distan de serlo, pues esta cuestión fue tomando forma de las manos de Shinya Yamanaka y Bertran Gurdon, cuando Yamanaka descubrió cuatro factores clave que podían inducir la reprogramación celular: Oct4, Sox2, Klf4 y c-Myc, los cuales recibieron el nombre de "factores de Yamanaka", logrando así en 2006 el hito de generar células madre pluripotentes inducidas (iPS) a partir de células diferenciadas humanas, y posteriormente en 2012, ambos fueron galardonados con el Premio Nobel por sus descubrimientos sobre la reprogramación celular (3,4,5).

Figura 3. Yamanaka y Gurdon
Según explicó Yamanaka en una entrevista, el desarrollo de las iPS fue consecuencia de la conjunción de diversas corrientes de investigación, entre las cuales cabe destacar: la reprogramación celular por transferencia nuclear, iniciada por John Gurdon en 1962; el descubrimiento de los factores de transcripción a partir de los experimentos de Schneuwly y sus colaboradores desde 1987; y la tercera los trabajos de aislamiento y cultivo de células troncales, de origen embrionario entre 1981 y 1998 (3,5).
Este descubrimiento (las iPS), supuso un gran avance, pues abrió las puertas a una cantidad insospechada de posibilidades hasta el momento, como es el estudio de enfermedades incurables y el efecto de los fármacos, e investigar las diversas posibilidades de trasplantes autólogos de células.

Figura 4. Evolución iPS
(Click en la imagen para aumentar tamaño)
En la actualidad, Yamanaka y su equipo están investigando cómo mejorar la eficiencia y la seguridad de la generación de células iPS, así como el uso de estas células en el tratamiento de enfermedades como la diabetes, el cáncer y la enfermedad de Parkinson.
Por su parte, Gurdon es conocido por sus investigaciones pioneras en la diferenciación celular y la reprogramación nuclear, que llevaron al descubrimiento que todas las células del cuerpo contienen el mismo ADN y que la información genética puede ser reprogramada para producir cualquier tipo de célula del cuerpo.
En estos momentos, Gurdon y su equipo están estudiando cómo se controla la expresión de los genes durante la diferenciación celular, y cómo se pueden utilizar las células madre y la reprogramación celular para el tratamiento de enfermedades como la enfermedad de Alzheimer y la enfermedad de Huntington.
En cuanto a las posibilidades futuras de la investigación de Yamanaka y Gurdon, se espera que sus avances en la generación de células y tejidos específicos para trasplantes, así como en la reprogramación celular, puedan llevar a importantes avances en la medicina regenerativa y el tratamiento de enfermedades actualmente incurables. Además, sus investigaciones también podrían tener aplicaciones en otras áreas, como la biotecnología y la agricultura.
Un enfoque prometedor es la reprogramación celular in vivo, que consiste en inducir la reprogramación celular en el propio cuerpo del paciente. Esto se puede lograr mediante la administración de factores de Yamanaka o la activación de vías de señalización asociadas con la reprogramación celular.

Figura 5. Esquema de la generación de células iPS
(Click en la imagen para aumentar tamaño)
Esta reversión celular puede tener aplicaciones en el tratamiento de enfermedades degenerativas asociadas con el envejecimiento, como la enfermedad de Alzheimer, la enfermedad de Parkinson y la enfermedad de Huntington. Además, también puede tener aplicaciones en la regeneración de tejidos y órganos dañados por el envejecimiento o la enfermedad.
Estos factores pueden ser administrados directamente en el cuerpo del paciente utilizando vectores virales o proteínas. Una vez dentro de las células, los factores de reprogramación activan vías de señalización específicas que inducen la reprogramación celular, como la vía de señalización Wnt, la vía de señalización Notch y la vía de señalización de la proteína morfogenética ósea (BMP) (4,6).
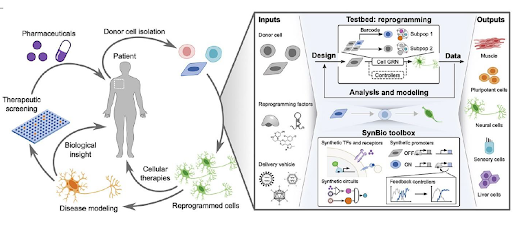
Figura 6. Resumen Proceso Reprogramación Celular
(Click en la imagen para aumentar tamaño)
APLICACIONES EN HEMATOLOGÍA
La reprogramación celular mediante factores de Yamanaka ha sido utilizada para generar células hematopoyéticas a partir de iPSs, lo que ofrece nuevas opciones terapéuticas para el tratamiento de enfermedades hematológicas. Por ejemplo, la anemia de células falciformes y la beta-talasemia son enfermedades genéticas de la sangre que pueden ser tratadas mediante terapia celular con células hematopoyéticas modificadas genéticamente. La terapia celular basada en iPSs reprogramadas con factores de Yamanaka ofrece una fuente ilimitada de células hematopoyéticas para el tratamiento de estas enfermedades.
Además, los factores de Yamanaka también se han utilizado en la investigación de enfermedades hematológicas, como la leucemia, para generar células madre pluripotentes inducidas de pacientes con la enfermedad. Estas células pueden ser diferenciadas en células hematopoyéticas afectadas por la enfermedad, lo que permite a los investigadores estudiar la patogénesis de la enfermedad y probar nuevas terapias.
Sin embargo, hay algunos desafíos que deben abordarse antes de que la terapia celular basada en iPSs pueda convertirse en una terapia convencional para enfermedades hematológicas. Uno de los mayores desafíos es el riesgo de la formación de tumores, ya que las iPSs tienen un mayor potencial tumorogénico que las células madre hematopoyéticas adultas. Por lo tanto, es importante continuar investigando y desarrollando técnicas de diferenciación y purificación de células que puedan reducir el riesgo de la formación de tumores.
Otro desafío importante es el costo y la disponibilidad de las iPSs para la terapia celular. La producción de iPSs es costosa y requiere una gran cantidad de tiempo y recursos. Además, hay un gran desafío en la creación de un banco de iPSs que sea compatible con la mayoría de la población, ya que cada persona tiene un perfil genético único (4,7).
Sin embargo, la reprogramación celular in vivo también plantea desafíos y preocupaciones éticas a parte de los mencionados. Pues a parte de las dificultades técnicas y aquellas que podrían considerarse éticas o morales, existen otra serie de aspectos a tener muy en cuenta en relación con la privacidad y la seguridad de los datos genómicos del paciente. Ya que el proceso in vivo implica la manipulación genómica de las células del paciente, lo que puede resultar en la recolección de datos genómicos y la posible exposición de información personal y sensible. Por lo tanto, es importante establecer medidas de seguridad y privacidad adecuadas para proteger los datos genómicos del paciente y garantizar su confidencialidad (8).
A pesar de estos desafíos, el futuro de la hematología y de la medicina regenerativa se ven prometedoras con la generación de células madre pluripotentes inducidas y la terapia celular basada en estas células. La tecnología seguirá evolucionando y mejorando, lo que permitirá un tratamiento más seguro y efectivo para enfermedades hematológicas.
En conclusión, los factores de Yamanaka han revolucionado la investigación en estos campos, teniendo un gran potencial para proporcionar nuevas soluciones en el tratamiento de enfermedades hasta la fecha consideradas incurables o de gran dificultad. Sin embargo, es importante abordar cuidadosamente los desafíos y preocupaciones éticas asociados con la reprogramación celular in vivo antes de su aplicación clínica.
OTRAS LÍNEAS DE INVESTIGACIÓN
La parabiosis es una técnica experimental que consiste en la unión quirúrgica de dos animales, generalmente ratones, de diferentes edades y/o genotipos, con el objetivo de estudiar la interacción entre sus sistemas circulatorios.
Esta también ha sido utilizada en la investigación en hematología para estudiar la interacción entre células sanguíneas y su microambiente. En particular, la técnica ha permitido el estudio de las células madre hematopoyéticas y su nicho, así como el papel de los factores circulatorios en la regulación de la hematopoyesis. Ya que en los inicios de la técnica se observaba que había algo en la sangre que al conectar ambos sistemas circulatorios actuaba positivamente sobre el individuo envejecido, pero todavía no se sabía cuál era este elemento.
Uno de los estudios más destacados en este campo fue realizado por los investigadores Rossi y colaboradores en 2012, quienes llevaron a cabo una parabiosis entre ratones jóvenes y viejos para estudiar el impacto del envejecimiento en las células madre hematopoyéticas y su capacidad para regenerar el sistema sanguíneo. Los resultados del estudio mostraron que la exposición de células madre hematopoyéticas de ratones jóvenes al ambiente circulatorio de ratones viejos resultó en una disminución en su capacidad para regenerar el sistema sanguíneo.
Estos hallazgos proporcionaron información importante sobre la regulación de la hematopoyesis y el impacto del envejecimiento en las células madre hematopoyéticas, lo que puede tener implicaciones para el desarrollo de tratamientos para enfermedades sanguíneas relacionadas con el envejecimiento.
Así, tras varios estudios, finalmente en el ensayo de Loffredo et al. (2013) se identificó al GDF11 (growth differentiation factor 11) como una molécula cuya concentración aumenta en la sangre de los ratones viejos al ser sometidos a parabiosis con ratones jóvenes. Esta molécula está relacionada con la hormona del crecimiento, se expresa en muchos tejidos (en gran medida en el bazo) y su producción declina con la edad.
Asimismo, se ha descubierto que GDF11 está involucrado en la regulación de la hematopoyesis, demostrándose que la administración de GDF11 aumenta la producción de células sanguíneas en animales de laboratorio, lo que sugiere que podría tener un potencial terapéutico en trastornos hematológicos, como la anemia (9,10).

Figura 7. cambios morfológicos y funcionales asociados a GDF11
(Click en la imagen para aumentar tamaño)
Sin embargo, la investigación sobre el papel de GDF11 en la hematología aún se encuentra en sus primeras etapas y se necesitan más estudios para comprender completamente su función y potencial terapéutico en enfermedades hematológicas en humanos.
En resumen, la parabiosis se presenta como una herramienta valiosa en la investigación en hematología, permitiendo el estudio de la interacción entre células sanguíneas y su microambiente, lo que puede tener implicaciones importantes en el desarrollo de tratamientos para enfermedades sanguíneas relacionadas con el envejecimiento, así como la participación de GDF11 en varios de los procesos fisiológicos.
REJUVENECIMIENTO MULTIÓMICO DE CÉLULAS HUMANAS
Este es un tema de investigación que ha despertado un gran interés en la comunidad científica en los últimos años. Esta técnica consiste en la aplicación de tratamientos para revertir el envejecimiento de las células a nivel molecular y celular, permitiendo que las células vuelvan a su estado más joven y saludable (11).
La clave del rejuvenecimiento multiómico está en la reprogramación de las células a través de la manipulación de su epigenoma, es decir, las modificaciones químicas que se realizan en el ADN para controlar la expresión génica. Al modificar el epigenoma de las células, se pueden revertir los cambios asociados al envejecimiento, como la acumulación de daño en el ADN y las mutaciones genéticas.

Figura 8. Reprogramación celular, la misma célula rejuvenece
(Click en la imagen para aumentar tamaño)
Uno de los enfoques más prometedores en el rejuvenecimiento multiómico es la utilización de factores de transcripción para reprogramar las células. Los factores de transcripción son proteínas que se unen al ADN y regulan la expresión de los genes. Al introducir factores de transcripción específicos en las células, es posible reprogramar su epigenoma y revertir el proceso de envejecimiento.
La terapia de rejuvenecimiento multiómico también puede tener aplicaciones en el campo de la medicina regenerativa. Al rejuvenecer las células de un paciente, es posible mejorar su capacidad de regeneración y curación, lo que puede ser especialmente beneficioso en el tratamiento de enfermedades crónicas y lesiones (12,13).
Sin embargo, aún existen varios desafíos técnicos y éticos (8) que deben ser abordados antes de que la terapia de rejuvenecimiento multiómico pueda ser ampliamente utilizada en la práctica clínica. Por ejemplo, aún se desconocen los efectos a largo plazo de la reprogramación celular, y existen preocupaciones sobre la posible aparición de mutaciones genéticas y el riesgo de cáncer.
En conclusión, la terapia de rejuvenecimiento multiómico de células humanas es un tema de investigación fascinante que tiene el potencial de revolucionar la medicina regenerativa y el tratamiento de enfermedades relacionadas con el envejecimiento. Si bien aún quedan muchas preguntas por responder, la investigación en este campo continúa avanzando y es probable que surjan nuevas terapias innovadoras en el futuro.
RELACIÓN ENTRE TERAPIA GÉNICA Y HEMATOLOGÍA
Dentro del ámbito de la salud, la medicina regenerativa es una disciplina emergente que busca desarrollar terapias innovadoras para reparar o reemplazar tejidos dañados o enfermos. La hematología, por su parte, es una rama de la medicina que se enfoca en el estudio y tratamiento de trastornos de la sangre, como anemia, leucemia, linfoma y trastornos de coagulación (11).
No obstante, como se puede deducir tras lo expuesto en esta entrada anteriormente, existe una estrecha relación entre la medicina regenerativa y la hematología, ya que muchas enfermedades hematológicas pueden ser tratadas mediante la regeneración de células sanguíneas saludables. En este sentido, la terapia de células madre y la terapia génica son dos formas de medicina regenerativa que se utilizan en la hematología para reemplazar las células sanguíneas enfermas o dañadas.
La terapia de células madre consiste en la introducción de células madre saludables en la médula ósea del paciente para reemplazar las células sanguíneas enfermas o dañadas. La terapia génica, por su parte, implica la introducción de genes sanos en las células sanguíneas del paciente para corregir mutaciones genéticas que causan enfermedades de la sangre.
Además, la medicina regenerativa también puede utilizarse para desarrollar terapias para otros trastornos de la sangre, como la beta talasemia. Esta enfermedad se puede tratar mediante la introducción de genes sanos en las células sanguíneas del paciente para estimular la producción de hemoglobina.(12)

Figura 9. Medicina Regenerativa
En conclusión, la medicina regenerativa y la hematología están estrechamente relacionadas debido a la capacidad de la medicina regenerativa para reemplazar las células sanguíneas enfermas o dañadas en el cuerpo. La terapia de células madre y la terapia génica son dos formas de medicina regenerativa que se utilizan comúnmente en la hematología para tratar trastornos de la sangre, y es probable que surjan nuevas terapias regenerativas para tratar enfermedades de la sangre en el futuro.
CONCLUSIONES
Tras lo expuesto anteriormente en esta entrada, no sería descabellado llegar a pensar que realmente en un futuro sí podríamos llegar a ser como la Turritopsis dohrnii o quizás incluso algo similar a la película In Time, ya que si se pudiera manipular el tiempo de vida, no cabe duda que el ser humano lo legislaría de alguna forma como ocurre en el Film mencionado, o si no, a este nivel, quizás sí que podemos pensar en llegar a ser mucho más longevos de lo que actualmente es el ser humano, pero ¿cuándo podría llegar este futuro? ¿Realmente es algo futuro y lejano?
Titular Abril 2022: Un salto en el tiempo: una nueva técnica rebobina la edad de las células de la piel en 30 años (14).
Quizás todo está mucho más cerca de lo que parece.
BIBLIOGRAFÍA
1. Aragón H de. Qué hace inmortal a la medusa «Turritopsis dohrnii» [Internet]. heraldo.es. [citado 13 de marzo de 2023]. Disponible en: https://www.heraldo.es/noticias/aragon/2022/08/30/medusa-inmortal- 1596643.html
2. Conocimiento V al. Los animales campeones de la regeneración [Internet]. OpenMind. 2023 [citado 13 de marzo de 2023]. Disponible en: https://www.bbvaopenmind.com/ciencia/biociencias/animales-la- regeneracion/
3. InfoVeloz.com. Nobel de Medicina 2012 para John Gurdon y el japonés Shinya Yamanaka [Internet]. InfoVeloz.com. [citado 13 de marzo de 2023]. Disponible en: https://www.infoveloz.com/post/nobel-de-medicina-2012-para-john- gurdon-y-el-japones-shinya-yamanaka_48459
4. Ortuño-Costela M del C, Cerrada V, García-López M, Gallardo ME. The Challenge of Bringing iPSCs to the Patient. International Journal of Molecular Sciences. enero de 2019;20(24):6305.
5. Shinya Yamanaka [Internet]. Premios Fronteras. [citado 13 de marzo de 2023]. Disponible en: https://www.premiosfronterasdelconocimiento.es/galardonados/shinya- yamanaka/
6. Epigenética de la transdiferenciación y reprogramación celular | Revista de la Sociedad Española de Bioquímia y Biología Molecular | SEEBM [Internet]. [citado 19 de marzo de 2023]. Disponible en: https://revista.sebbm.es/articulo.php?id=151&url=epigenetica-de-la-transdiferenciacion-y-reprogramacion-celular
7. Wang NB, Beitz AM, Galloway K. Engineering cell fate: Applying synthetic biology to cellular reprogramming. Current Opinion in Systems Biology. 1 de diciembre de 2020;24:18-31.
8. Gámez Escalona JA. Las IPS paradigma ético de la investigación biomédica. Cuadernos de Bioética. 2013;24(82):419-42.
9. Parabiosis y su combinación con otras técnicas para descubrir los secretos de la sangre. Pensar en Movimiento: Revista de Ciencias del Ejercicio y la Salud. 19 de febrero de 2014;12(1):1-15.
10. SAVALnet - La sangre joven rejuvenece [Internet]. SAVALnet. [citado 19 de marzo de 2023]. Disponible en: https://www.savalnet.cl/CienciayMedicina/ProgresosMedicos/la-sangre-joven- rejuvenece.html
11. Fernández-Rúa JM. Identifican el gen responsable del envejecimiento celular [Internet]. biotechmagazineandnews.com. 2020 [citado 29 de marzo de 2023]. Disponible en: https://biotechmagazineandnews.com/identifican-el-gen- responsable-del-envejecimiento-celular/
12. Elsevier. La medicina regenerativa: la cura del futuro (alzhéimer, párkinson, cáncer,…) [Internet]. Elsevier Connect. [citado 29 de marzo de 2023]. Disponible en: https://www.elsevier.com/es-es/connect/ciencia/la-medicina-regenerativa- la-cura-del-futuro-alzheimer-parkinson-cancer
13. Gill D, Parry A, Santos F, Okkenhaug H, Todd CD, Hernando-Herraez I, et al. Multi- omic rejuvenation of human cells by maturation phase transient reprogramming. Tyler JK, editor. eLife. 8 de abril de 2022;11:e71624.
14. Titular: Un salto en el tiempo: una nueva técnica rebobina la edad de las células de la piel en 30 años: A jump through time – new technique rewinds the age of skin cells by 30 years | Babraham Institute [Internet]. [citado 3 de abril de 2023]. Disponible en: https://www.babraham.ac.uk/news/2022/04/new-technique- rewinds-age-skin-cells-30-years
REFERENCIAS DE LAS IMÁGENES
- Figura 1: 32497116_1655904308638.png (1416×1200) [Internet]. [citado 13 de marzo de 2023]. Disponible en: https://images.jifo.co/32497116_1655904308638.png
- Figura 2: BBVA-OpenMind-Yanes-Los-animales-campeones-de-la- regeneracion_1.webp (572×380) [Internet]. [citado 13 de marzo de 2023]. Disponible en: https://www.bbvaopenmind.com/wp- content/uploads/2023/02/BBVA-OpenMind-Yanes-Los-animales-campeones-de-la- regeneracion_1.webp
- Figura 3: 1563496202_380684.webp (722×442) [Internet]. [citado 13 de marzo de 2023]. Disponible en: https://www.infoveloz.com/storage/12/10/08/06/fit722x442/1563496202_38068 4.webp
- Figura 4: ijms-20-06305-g001.png (3296×1768) [Internet]. [citado 13 de marzo de 2023]. Disponible en: https://www.mdpi.com/ijms/ijms-20- 06305/article_deploy/html/images/ijms-20-06305-g001.png
- Figura 5: 179_p09a.jpg (500×252) [Internet]. [citado 19 de marzo de 2023]. Disponible en: https://revista.sebbm.es/imagenes/179_p09a.jpg
- Figura 6: 1-s2.0-S2452310020300342-fx1.jpg (497×200) [Internet]. [citado 19 de marzo de 2023]. Disponible en: https://ars.els-cdn.com/content/image/1-s2.0- S2452310020300342-fx1.jpg
- Figura 7: 336765pi001.jpg (650×454) [Internet]. [19 de marzo de 2023]. Disponible en: https://www.savalnet.cl/medios/cyc/progresos/2014/336765pi001.jpg
- Figura 8: envejecimiento.jpg (1200×800) [Internet]. [citado 29 de marzo de 2023]. Disponible en: https://biotechmagazineandnews.com/wp- content/uploads/2020/11/envejecimiento.jpg
- Figura 9: medicina-regenerativa.jpg (874×507) [Internet]. [citado 29 de marzo de 2023]. Disponible en: https://www.elsevier.com/ data/assets/image/0014/602231/medicina- regenerativa.jpg
‘ Medice, cura aegrotum, sed non morbum’L.A. Séneca, filósofo romano, 4 a. C.-a 65 d. C.
Cada vez que sientas que no le importas a nadie, recuerda que, tan sólo en un milímetro de sangre tienes alrededor de 11 millones de leucocitos que darían la vida por ti con tal de protegerte.