Autor : Rubén M. F.
4º Curso de Medicina grupo B (curso 2021-22)
Código de trabajo : 2110-RMF
INTRODUCCIÓN
Las leucemias son un grupo de neoplasias clonales que surgen de la transformación maligna de células hematopoyéticas, acumulándose células anormales en la médula ósea, sangre periférica y que también infiltra órganos.
Las leucemias pueden ser agudas o crónicas (según el grado de diferenciación celular). Éstas, a su vez, según la línea celular proliferante, se dividen en linfoides y mieloides. En este caso, trataremos las leucemias agudas linfoblásticas (LLA), las cuales son proliferaciones clonales malignas de células progenitoras o precursoras linfoides en distintos grados de diferenciación.
Representan el cáncer más frecuente en la edad pediátrica (infancia y adolescencia con pico de incidencia entre 2-5 años), concretamente el 80%, siendo el 85% LLA de precursores B y el 15% de estirpe T.
En cuanto al sexo, la LLA predomina ligeramente en los varones, sobre todo en la edad puberal. Las diferencias geográficas son notables en esta enfermedad; mientras que, en los países menos desarrollados, como Norte de África y Oriente Medio, predominan los linfomas y las LLA de estirpe T, en los países industrializados la LLA de estirpe B es, con diferencia, la más frecuente de las hemopatías malignas (1). Este hecho se ha relacionado con la mayor facilidad para la exposición a determinados agentes medioambientales “leucemógenos” en los países industrializados. En los países con poblaciones heterogéneas, se ha observado una mayor incidencia de LLA en la raza blanca.
Asimismo, los pacientes con Síndrome de Down tienen más probabilidades de desarrollar LLA.
FISIOPATOLOGÍA
La LLA es causada por una serie de aberraciones genéticas y moleculares adquiridas. Por ejemplo, en el caso del LLA de estirpe B, encontramos traslocaciones comunes como t(12;21) [ETV6–RUNX1](25%), t(1;19) [TCF3– PBX1](5%), t(9;11) [BCR–ABL1](3%), así como traslocaciones que incluyen al gen MLL. También encontramos hiperploidía (>50 cromosomas) (25%), con buen pronóstico, e incluso hipodiploidía (<44 cromosomas), aunque menos frecuente (2-3%) y de pronóstico desfavorable. Ciertos cambios genéticos se asocian a implicaciones pronósticas y terapéuticas (2).
Tabla 1. Anomalías estructurales. Alteraciones citogenéticas y moleculares (3)
Los estudios de NGS (Next-generation Secuencing) están ayudando a comprender e incrementar la capacidad de identificar alteraciones genéticas submicroscópicas y secuenciar mutaciones en la LLA, lo que ayudará a definir nuevas alteraciones moleculares. Por ejemplo, la delección IKZF1 predice un pobre pronóstico. Todo ello conllevará implicaciones pronósticas y/o como posibles dianas terapéuticas.
CLASIFICACIÓN
Según la clasificación morfológica del grupo cooperativo Franco-Americano-Británico podemos distinguir tres subtipos de LAL, designados como L1, L2 y L3.
Tabla 2. Clasificación FAB (3).
Figura 1. Clasificación del grupo cooperativo Franco – Americano – Británico de las leucemias agudas linfoblásticas: L1, L2 y L3.(3)
Tabla 3. Clasificación inmunológica de la LLA.(3)
DIAGNÓSTICO
En principio, se debe realizar una anamnesis y exploración física completas, donde anotaremos los principales síntomas de nuestro paciente entre los cuales, se encuentran la fiebre (60%), palidez (55%), petequias y equimosis (50%), dolores osteoarticulares (25%), astenia y anorexia (33%), así como hepatoesplenomegalia (65%) y adenopatías (60%), secundarios a la infiltración celular. Es importante la realización de una exploración testicular en varones, así como una exploración neurológica exhaustiva para descartar síntomas secundarios a la infiltración celular.
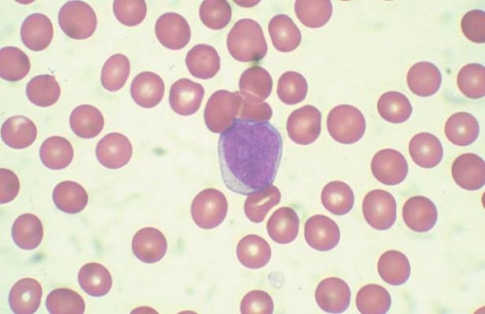
Los estudios analíticos nos mostrarán anemia (85%), trombopenia general moderada (70%), leucocitosis > 10x10^9 /L (50%), neutropenia < 0.5x10^9 /L (40%). Incluso podemos observar elevación de LDH, fósforo y ácido úrico en aquellos con elevada carga leucémica. Por otra parte, en el frotis periférico, la pancitopenia y los blastos periféricos (Figura 2) sugieren una leucemia aguda, y a diferencia de lo que ocurre en la LMA, los bastones de Auer (inclusiones azurofílicas lineales en el citoplasma de las células blásticas) nunca están presentes en la leucemia linfoblástica aguda.
El diagnóstico definitivo de una leucemia aguda siempre se debe realizar mediante el análisis morfológico, molecular y citogenético del aspirado de la médula ósea. Nunca deberemos iniciar un tratamiento sin haber obtenido una muestra de médula ósea. La presencia de al menos un 25% de blastos confirmará el diagnóstico.
Entre otras pruebas también encontramos la punción lumbar, para descartar la afectación del SNC, la radiografía de tórax, que nos permitirá conocer si hay afectación mediastínica (presentación común en la LLA-T, presente en niños mayores y asociada a hiperleucocitosis. Figura 1), así como más pruebas tales como la ecografía abdominal, testicular (si exploración patológica) estudio cardiológico (previo al uso de fármacos cardiotóxicos), bioquímica sanguínea (incluyendo LDH, ácido úrico, calcio, fósforo, transaminasas, etc.), estudio de coagulación, serologías (hepatitis viral, VIH, herpes, CMV, etc.) e inmunoglobulinas. Si el paciente presenta fiebre, se deben obtener cultivos de sangre, orina y de cualquier lesión sospechosa e iniciar el tratamiento antibiótico adecuado.(4)
Figura 2. Blasto en sangre periférica.(5)
PRONÓSTICO
Son factores de buen pronóstico la LAL pre-B o B común, la presencia de <5% de blastos en médula ósea tras 2 semanas de tratamiento y la hiperdiploidía (>50 cromosomas). Mientras tanto, se consideran factores de mal pronóstico:
- Edad: niños <1 y >10 años, adultos >30 años.
- Sexo masculino.
- Presencia de adenopatías, masas o visceromegalias.
- Infiltración del sistema nervioso central.
- Leucocitos >50.000/mm3
- Inmunofenotipo: LAL-B no común o CALLA negativa (CD10-), LAL-T no cortical.
- Alteraciones citogenéticas: t (9,22) – cromosoma Filadelfia- o reordenamiento bcr-abl, 11q23, t(4,11), hipodiploidía (<46 cromosomas).
- Mutación IKAROS.
- Respuesta lenta al tratamiento: >10% de blastos en médula ósea tras 2 semanas de tratamiento o ausencia de respuesta tras 4-5 semanas de tratamiento.(6)
Atendiendo a los factores pronósticos, estos grupos de pacientes podrían dividirse en tres apartados (4):
|
Riesgo
estándar
(25%)
|
Riesgo
alto
(12-15%)
|
|
Todos
los
siguientes
criterios:
|
Uno
o
más
de
los
siguientes:
|
-
Leucocitos
<20x109/L
al
diagnóstico
-
Inmunofenotipo
no
T
-
Ausencia
de
infiltración
del SNC
y/o testes
-
Citogenética
(uno
de
los
criterios
es
suficiente):
-
No
t(1;19)
-
No
reordenamiento
MLL
-
Presencia
de
<1.000
blastos/mm3
en
día
+8
de
la
inducción,
en
sangre
periférica
-
Presencia
de <5%
de blastos y <0,1%
de
ERM
en
médula
ósea (MO)
en día
+15 de la
inducción
y
al
final
de
la inducción
IA
|
-
ERM
>1% en el día +33 de la
inducción
en la
médula ósea
-
ERM
>0.1%
antes
de
la
consolidación,
en
médula
ósea
|
|
Riesgo
intermedio
(60%)
|
|
Todos
los
restantes
|
TRATAMIENTO
En general comprende: inducción de la remisión, consolidación/intensificación, reinducción y mantenimiento. Con los protocolos actuales, generalmente se obtienen tasas de supervivencia libre de enfermedad en primera remisión completa del 70-80% (4).
Inducción de la remisión
En esta fase, se incluyen los corticoides, así como vincristina y al menos un tercer agente (asparraginasa o daunorrubicina o ambos). Incluso el grupo BFM intensifica esta fase con una segunda inducción con citarabina a bajas dosis, CFM y mercaptopurina. Asimismo, esta fase requiere un tratamiento de soporte importante, mediante prevención de lisis tumoral, manejo de complicaciones metabólicas, profilaxis y tratamiento de las infecciones.
Consolidación/Intensificación
En bajo riesgo e intermedio, empleamos metotrexate a altas dosis con mercaptopurina.
En alto riesgo, intensificamos en forma de bloques de citostáticos a altas dosis y combinados.
Reinducción
En las LLA de riesgo bajo e intermedio, el esquema consiste en repetir un régimen similar a la inducción a los tres meses de la remisión completa.
Mientras tanto, en las LLA de alto riesgo, depende según el caso. En aquellos pacientes que no son candidatos a TPH se continúa con bloques de reinducción intensiva antes del mantenimiento. Sin embargo, si hay indicación de TPH, no se realiza la reinducción
Mantenimiento
Los niños requieren un tratamiento prolongado. Además, los pacientes sin indicación de TPH reciben tratamiento mantenimiento hasta completar al menos 24 meses de quimioterapia. El tratamiento consiste en la combinación de mercaptopurina oral diaria con metotrexato oral semanal.
Profilaxis y tratamiento sobre SNC
Clasificación de afectación del SNC (SEHOP-PETHEMA 2013):
- SNC-1: ausencia de blastos en el LCR
- SNC-2 blastos en LCR con <5 leucocitos/µL y/o punción lumbar traumática (>10 eritrocitos) o hemorragia, con blastos.
- SNC-3: blastos en el LCR con >5 leucocitos/µL y/o afectación de pares craneales y/o masa tumoral en cerebro o meninges, detectada por imagen.
Se utiliza quimioterapia sistémica a altas dosis junto con tratamiento intratecal (metotrexato, citarabina e hidrocortisona), según edad entre 15-26 dosis según el grupo de riesgo.
Indicaciones de TPH (SEHOP-PETHEMA 2013) (7)
- No remisión completa citomorfológica tras la inducción A (día +33).
- Enfermedad mínima residual (ERM) >1% tras la inducción A y ERM >0,1% en el día +78.
- En t(4;11) con ERM >0,1% en el día +78
- En hipodiploidía (<44 cromosomas) con ERM >0,1% en el día +78
- En LLA-T con mala respuesta a prednisona y con ERM >0,1% en el día +78.
- En pacientes de alto riesgo si la ERM es persistentemente positiva >0,01% (tras tercer bloque AR-3)
Recaídas
Un 15-20% de los pacientes recaen. Podemos establecer una serie de factores pronósticos de recaída tales como el tiempo transcurrido desde el final del tratamiento hasta la recaída, el cual es peor si es <6 meses, la localización de la recaída, con mejor pronóstico si es extramedular aislada, el inmunofenotipo, con peor pronóstico en las recaídas LLA-T…
El tratamiento se basa en poliquimioterapia y TPH en algunos casos. Todo ello ofrece unos resultados del 40% de rescates aproximadamente.
Nuevos tratamientos en la LLA
Cabe destacar la importancia de la progresiva aparición de nuevos tratamientos en la LLA tales como anticuerpos monoclonales (inotuzumab ozogamicina –anti- CD22 y blinatumumab-anti-CD19 BITE, inhibidores de la tirosincinasa y rituximab -anti-CD20), inhibidores del proteasoma, moduladores epigenéticos, terapias moleculares dirigidas y la terapia celular CAR-T, mediante la cual, se desarrollan linfocitos autólogos modificados genéticamente para expresar un receptor antigénico quimérico que incluye un anti CD19 unido a un dominio de señal intracelular. Por ejemplo, tisangeleucel ha sido a probado para pacientes pediátricos y jóvenes de hasta 25 años con LLA-B refractaria, en recaída post- transplante, o en segunda recaída o posterior. Asimismo, en un ensayo clínico realizado en el Clínic y en el Hospital Sant Joan de Déu, el CAR-T ARI-0001 demostró ser seguro y eficaz, por lo que fue aprobado por la AEMPS (8).
BIBLIOGRAFÍA
1.- J. F. Margolin, K. R. Rabi, C. P. Steuber and D. G. Poplack, “Acute Lymphoblastic Leukemia,” In P. A. Pizzo and D. G. Poplack, Eds., Principles and Practice of Pediatric Oncology, Lippincott Williams & Wilkins, Philadelphia, 2011, pp. 518-565. - References - Scientific Research Publishing [Internet]. [cited 2022 Feb 26]. Available from: https://
www.scirp.org/(S(i43dyn45teexjx455qlt3d2q))/reference/ReferencesPapers.asp x?ReferenceID=843455
2.- Bhojwani D, Yang JJ, Pui CH. Biology of Childhood Acute Lymphoblastic Leukemia. Pediatr Clin North Am [Internet]. 2015 Feb 1 [cited 2022 Feb 24];62(1):47. Available from: /pmc/articles/PMC4250840/
4.- Manual Práctico de Hematología Clínica 2019: 9788488825278: Sanz, M. — Carreras, E. — Rovira, M. — Sanz, J. | axon.es [Internet]. [cited 2022 Feb 26]. Available from: https://axon.es/ficha/libros/9788488825278/manual-practico-de-hematologia-clinica- 2019
5.- CARR JH. CLINICAL HEMATOLOGY ATLAS. 2021;
7.- SEHH - Sociedad Española de Hematología y Hemoterapia [Internet]. [cited 2022 Feb 26]. Available from: https://
www.sehh.es/
8.- La AEMPS autoriza el CAR-T ARI-0001 del Hospital Clínic para pacientes con leucemia linfoblástica aguda - Agencia Española de Medicamentos y Productos Sanitarios [Internet]. [cited 2022 Feb 26].