Autora: Inés G. N.
Curso 4º de Medicina Grupo A (Curso 2024-2025)
Código de trabajo : 2402-IGN
INTRODUCCIÓN
Se trata de un trastorno de la capa superficial de los eritrocitos que hace que tengan una forma esférica, predisponiéndolos a hemólisis mecánica en el bazo (1). Es una de las causas más frecuentes de anemia hemolítica hereditarias y también se conoce como enfermedad de Minkowski-Chauffard. Se considera una anemia no microcítica regenerativa congénita
FISOPATOLOGÍA
Como sabemos, los eritrocitos tienen una forma bicóncava, esto es importante ya que es la característica responsable de la deformabilidad eritrocitaria. Esta es la habilidad de los eritrocitos para cambiar su forma bajo cierto nivel de estrés aplicado, sin que exista hemólisis. Esta es una propiedad importante ya que los eritrocitos deben cambiar de forma al pasar por la microcirculación (2).
La deformabilidad es una habilidad intrínseca del corpúsculo determinada por la geometría y las propiedades de la membrana del eritrocito.

Figura 1. Paso del hematíe por los capilares sinusoidales.
La forma del eritrocito también nos va a condicionar la vida media del mismo. La manera en la que el bazo discrimina entre eritrocitos sano y viejos es mediante la capacidad que tengan de pasar por los estrechos sinusoides esplénicos (3).
Aquellas células que no tengan la flexibilidad suficiente como para pasar a través de las sinusoides se quedarán dentro de la pulpa roja, una zona rica en macrófagos que fagocitarán estas células.
Una vez visto las repercusiones de la perdida de elasticidad, vamos a explorar más profundamente las razones de que esto suceda en los esferocitos.
Los eritrocitos tienen una bicapa lipídica con unas proteínas integrales de membrana embebidas en ella. Actúan entre sí para crear una malla en la cara interna de la bicapa responsable de mantener la estabilidad y la flexibilidad (4).

Figura 2. Proteinas de la membrana eritrocitaria
Entre ellas destacan (5) :
- Espectrina: principal responsable de la estabilidad de la maya proteica conta de 2 subunidades enrolladas entre sí. Cadena Alpha codificada por el gen SPTA1 en el cromosoma 1 y cadena beta codificada por el gen SPTB en el cromosoma 14
- Ankirina: papel importante al unir la cadena de espectrinas con la Banda 3 y la Banda 4.2. Codificada por el gen ANK1 en el cromosoma 8
- Banda 3: tiene 2 dominios e interviene activamente en la eliminación de eritrocitos envejecidos. Codificada por el gen SLC4A1 en el cromosoma 17
- Banda 4.2: modulador para estabilizar la unión Anakirina y Banda 3. Codificada por el gen EPB42 en el cromosoma 15.
Mutaciones en estos genes se consideran ser los responsables de la esferocitosis hereditaria al alterar la membrana eritrocitaria y su estabilidad y flexibilidad. Hay que mencionar, que hay un gran porcentaje en el que estas mutaciones son dominantes y otro porcentaje menor en el que son recesivas.
CLÍNICA
- Anemia hemolítica dada la destrucción prematura de los eritrocitos, por lo que el paciente presentará astenia y palidez mucocutánea
- Ictericia: la destrucción de eritrocitos genera un aumento en la bilirrubina indirecta que nos lleva a ese color amarillento de mucosas y conjuntivas
- Esplenomegalia: esto es un signo común de esta enfermedad ya que la hemólisis se realiza en el bazo, el paciente se puede quejar de molestias abdominales. Podemos encontrarnos una esplenomegalia masiva en algunos casos
- Litiasis biliares: a raíz de la acumulación de bilirrubina (6).
Hay diferentes niveles de severidad de la enfermedad:
- - ESH moderada: Estos pacientes tienen hemoglobina de 8-10 g/dL, recuentos reticulocitarios cercanos a 10% y bilirrubinemia de 2-3 mg/dL. Se ve tanto en formas dominantes como en recesivas.
- - ESH grave: La anemia es grave (hemoglobina < 8 g/dL) y puede implicar compromiso de vida, por lo que son transfusión-dependientes.
La morfología eritrocitaria suele mostrar, además de los característicos esferocitos, esferocitos con contornos irregulares y/o poiquilocitosis. Esta forma se ve principalmente en pacientes con herencia recesiva.
DIAGNÓSTICO
- Historia clínica y examen físico: veremos los síntomas típicos de la anemia mencionados anteriormente, pero sobre todo veremos ictericia y una esplenomegalia palpable
- Pruebas de laboratorio: solicitaríamos lo siguiente
- Un Hemograma completo: veremos una Hb baja, lo cual nos indica anemia, a su vez veremos una VCM normal o ligeramente elevada lo que nos habla de que es normocítica.
- Una Bioquímica completa: veremos una bilirrubina indirecta elevada por el metabolismo de la Hb tras la hemólisis y una LDH aumentada (7)
- Reticulocitos en sangre periférica: en este caso veríamos unos reticulocitos aumentados ya que hay un intento de corregir la anemia
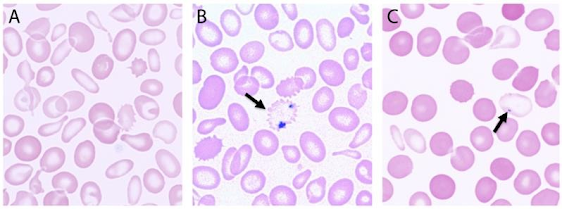
- Frotis de sangre; es la prueba clave ya que veremos unos eritrocitos esféricos

Figura 3. Esferocitos en frotis de sangre periférica
- Prueba de fragilidad osmótica: Es un examen de sangre para detectar si hay mayor probabilidad de que los glóbulos rojos se descompongan.Los glóbulos rojos se prueban con una solución salina hipotónica (8). Esto determina qué tan frágiles son, en esta patología la fragilidad osmótica estará aumentada (9).
- Prueba de eosina-5-malemida (EMA): Detecta anomalías en la estructura del citoesqueleto eritrocitario basándose en una unión específica de un colorante fluorescente, eosina 5´-maleimida (EMA), a la lisina 430 del dominio extracelular de la proteína banda 3. La disminución de la intensidad de fluorescencia emitida por EMA, medida por citometría de flujo, inferior al 75-80 % del valor normal indica disminución del contenido de esta proteína en los eritrocitos. Es una técnica moderna que podemos usar en esferocitos hereditaria para ver la deficiencia de esas proteínas de membrana en caso de que no se vea claro con las pruebas anteriores (10).
- Electroforesis de proteínas de la membrana eritrocitaria: Puede realizarse como paso previo al análisis genético para detectar la proteína afecta.
- Estudio Molecular de genes codificantes proteínas de membrana: Permite el diagnóstico de certeza en una membranopatía. Los defectos en las proteínas a-espectrina y proteína 4.2 son de carácter recesivo y pueden cursar con un fenotipo más agresivo.
- Pruebas de imagen: podríamos realizar una ecografía abdominal para valorar la presencia de litiasis y de esplenomegalia (7).
TRATAMIENTO
- Suplemento de ácido fólico: como hay hemólisis, el cuerpo intenta compensar esa pérdida produciendo más glóbulos rojos, damos ácido fólico para asegurar la producción correcta de eritrocitos y evitar que la anemia se torne hacia megaloblástica por excesiva necesidad de eritrocitos y bajos recursos para producirlos.
- Transfusión de sangre; podemos transfundirles en casos de anemia severa o durante crisis hemolíticas (7).
- Manejo quirúrgico
- ¿Cuándo hacemos esplenectomía?: lo hacemos en caso de:
- Anemia hemolítica grave que no responde a otros tratamientos.
- Crisis hemolíticas frecuentes que afectan a la calidad de vida del paciente.
- Presencia de complicaciones como litiasis biliares, molestias abdominales o infecciones recurrentes.
- Beneficios: con la esplenectomía estamos quitando el sitio principal de hemólisis, por ello disminuye la anemia, la hiperbilirrubinemia y el recuento de reticulocitos cae hasta cifras normales. Disminuimos las crisis hemolíticas y la necesidad de trasfusiones.
Además, en pacientes con molestias abdominales a causa de esplenomegalia paliaríamos los síntomas.
- Riesgos:
- Infecciones: es el riesgo mas relevante ya que estos pacientes son más susceptibles a padecer infecciones por bacterias encapsuladas, pudiendo llegar a una sepsis llamada síndrome séptico postesplenectomía.

Figura 4. Acción del bazo en la eliminación de bacterias encapsuladas.
El bazo constituye un excelente filtro para el aclaramiento de microorganismos, inmunocomplejos y hematíes parasitados. La producción esplénica de opsoninas, anticuerpos específicos y otros mediadores inmunes facilita el proceso de fagocitosis, especialmente de bacterias encapsuladas.El paso lento de la sangre a través de los sinusoides esplénicos y el contacto prolongado con el sistema reticuloendotelial, permite el tiempo suficiente para la eliminación bacteriana en ausencia de niveles elevados de anticuerpos (11). En orden de importancia tendríamos a:
- S.Pneumoniae,
- N.meningitidis
- H.influenzae
- Asociados a la cirugía: riesgo de hemorragia y de trombosis.
Vacunación: por lo general administramos vacunas contra las 3 bacterias mencionadas anteriormente :
- S. Pneumoniae; la vacuna conjugada 8 meses antes y la vacuna polisacárida 2 meses después. Revacunación con la polisacárida a los 5 años
- N. Meningitidis: la vacuna polisacárida 2 semanas antes de la cirugía. Revacunación con la polisacárida a los 5 años.
- H. Influenzae tipo B: 2 semanas antes de la cirugía (12).
A su vez damos antibióticos profilácticos antes y después de la cirugía y reforzamos las vacunas también después de la cirugía.
En niños menores de 6 años no indicamos esplenectomía dado un mayor riesgo de infección por estas bacterias, intentamos retrasar la esplenectomía hasta la completa maduración del sistema inmune.
PRONÓSTICO Y COMPLICACIONES
- Pronóstico: el pronóstico en general es bueno, un caso más complejo serían los niños en los que nos planteamos esplenectomía como última opción y debemos agotar los otros recursos terapéuticos primero.
- Complicaciones:
- Crisis aplásica: Son menos frecuentes que las crisis hemolíticas, pero de mayor gravedad. En pacientes con factores predisponentes como trastornos de la serie eritroide, hasta el 70-80% de las crisis aplásicas son debidas a infecciones por parvovirus B19, al tener tropismo por los precursores eritroides (7).
- Eritropoyesis extramedular: Esta complicación puede observarse en pacientes que no han sido esplenectomizados y se quita con la esplenectomía (7).
- Sobrecarga férrica: debido a un aumento de la absorción del Fe y secundario a transfusiones repetidas.
- Litiasis biliares
- Infecciones por bacterias encapsuladas en caso de esplenectomía, riesgo mayor en los primeros 2 años.
CONCLUSIÓN
La esferocitosis hereditaria causa una anemia hemolítica normocítica regenerativa congénita en la que hay una alteración de la membrana eritrocitaria.
Hemos aprovechado esta patología para explicar la composición de la membrana eritrocitaria y el funcionamiento de la fagocitosis de eritrocitos en el bazo.
También concluimos hablando de la esplenectomía como proceso curativo, pero a su vez de los numerosos riesgos que conlleva y la razón de estos.
A nivel personal quería comentar que la primera paciente que he visto nunca en un quirófano fue una niña de 15 años con esferocitosis hereditaria a la cual le extirparon un bazo de 23 cm, refería astenia y grandes molestias abdominales a causa de esta esplenomegalia masiva. En su día me pareció fascinante ver un caso así, y tiempo después volvimos a ver a la paciente, la cual no tenía anemia, ictericia ni molestias abdominales y a la cual le había mejorado mucho la calidad de vida la esplenectomía realizada.
BIBLIOGRAFÍA
1. Anemia esferocítica hereditaria (internet). Disponible en: https://medlineplus.gov/spanish/ency/article/000530.htm
2. Deformabilidad eritrocitaria (internet). Disponible en:
https://es.wikipedia.org/wiki/Deformabilidad_eritrocitaria
3. Eriptosis (internet). Disponible en :https://www.kenhub.com/es/library/anatomia-es/eritrocitos
4. Aspectos bioquímicos de la esferocitosis hereditaria (internet). Disponible en:
http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-02892002000100001
5. Genes implicados en esferocitosis hereditaria (internet). Disponible en:
https://www.ivami.com/es/pruebas-geneticas-mutaciones-de-genes-humanos-enfermedades-neoplasias-y-farmacogenetica/1357-pruebas-geneticas-esferocitosis-hereditaria-hereditary-spherocytosis-genes-i-ank1-epb42-slc4a1-spta1-i-y-i-sptb
6. Manifestaciones clínicas de la esferocitosis hereditaria (Artículo). Disponible en:
Chrome-extension://efaidnbmnnnibpcajpcglclefindmkaj/https://www.sap.org.ar/uploads/consensos/esferocitosis-hereditaria-revisi-oacuten-parte-ii-manifestaciones-cl-iacutenicas-evoluci-oacuten-complicaciones-y-tratamiento.pdf
7. Tratamiento de la esferocitosis (artículo): Disponible en:
chrome-extension://efaidnbmnnnibpcajpcglclefindmkaj/https://www.sehop.org/wp-content/uploads/2022/04/GUIA-ESFEROCITOSIS-HEREDITARIA-SEHOP.pdf
8. Prueba de fragilidad osmótica (internet). Disponible en:
https://www.cibic.com.ar/laboratorios-bioquimicos/evaluacion-fragilidad-osmotica-los-hematies-citometria-flujo/
9. Prueba de fragilidad osmótica (internet). Disponible en: https://medlineplus.gov/spanish/ency/article/003641.htm
10. Prueba de EMA (internet). Disponible en:
https://empendium.com/manualmibe/tratado/chapter/B76.VI.C.2.2.6.
11. Susceptibilidad a la infección por bacterias encapsuladas en pacientes esplenectomizados. (Artículo). Disponible en:
chrome-extension://efaidnbmnnnibpcajpcglclefindmkaj/file:///C:/Users/Usuario/Downloads/S0009739X01718271.pdf
12. Protocolo de vacunación post esplenectomía. (archivo pdf). Disponible en:
chrome-extension://efaidnbmnnnibpcajpcglclefindmkaj/https://www.serviciofarmaciamanchacentro.es/images/stories/recursos/recursos/protocolo/cirugia/vacunacion%20pacientes%20esplenectomizados_2013.pdf
chrome-extension://efaidnbmnnnibpcajpcglclefindmkaj/file:///C:/Users/Usuario/Downloads/S0009739X01718271.pdf
12. Protocolo de vacunación post esplenectomía. (archivo pdf). Disponible en:
chrome-extension://efaidnbmnnnibpcajpcglclefindmkaj/https://www.serviciofarmaciamanchacentro.es/images/stories/recursos/recursos/protocolo/cirugia/vacunacion%20pacientes%20esplenectomizados_2013.pdf