4º Curso Medicina grupo D (curso 2021-2022)
Código trabajo : 2117-MHM

Hipótesis del segundo hit:
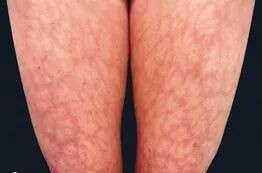
Ilustración 2. Lívedo Reticularis

INTRODUCCIÓN
El síndrome antifosfolípido es un trastorno autoinmunitario que cursa con trombofilia como signo principal. Se caracteriza por la coexistencia de trombosis, que pueden ser venosas o arteriales y/o pérdidas fetales recurrentes; a la vez que en sangre se encuentran anticuerpos antifosfolípidos circulantes, sobretodo los denominados anticoagulante lúpico, anticuerpos anticardiolipina o anti-b2 glicoproteína I.
Fue descrita por primera vez por el Dr. Graham RV Hughes, en 1983, y es considerada una de las causas más frecuentes de trombofilia adquirida y de accidentes cardiovasculares en menores de 50 años.
Ilustración 1. Dr. Graham Hughes
Su espectro clínico es muy amplio, y puede afectar a cualquier órgano o sistema, aunque las implicadas con mayor frecuencia son las venas profundas de los miembros inferiores y las arterias cerebrales.
EPIDEMIOLOGÍA
Los anticuerpos aFL, aparecen en el 1-5% de la población sana, siendo mayor el porcentaje cuanto más aumenta el rango de edad; la incidencia del SAF es de 5 por cada 100000 habitantes/año, siendo al prevalencia de 20-50 por cada 100000. Es más frecuente en mujeres, especialmente en el caso de asociación con Lupus Eritematoso Sistémico, en el que el ratio es de 1 hombre por cada 7 mujeres.
ETIOPATOGENIA
Los anticuerpos aFL son un grupo heterogéneo de inmunoglobulinas que se dirigen contra proteínas plasmáticas de unión a fosfolípidos o cofactores, fosfolípidos de membrana o complejos de unión. La activación de la célula endotelial por la unión de los anticuerpos genera la liberación de citoquinas y el metabolismo de prostaciclina.
Se supone como origen de los anticuerpos la exposición a agentes ambientales, como infecciones, en pacientes genéticamente predispuestos, por un proceso de mimetismo molecular; de esta forma se ha propuesto una asociación significativa entre la aparición de anticuerpos aFL y las trombosis en pacientes con infecciones por VEB, CMV, VHC, adenovirus o parvovirus.
Del mismo modo, ocurriría con fármacos o neoplasias, aunque se relacionan menos con trombosis y los anticuerpos están presentes de manera transitoria.
Los aFL activarían los monocitos y las células endoteliales con un aumento de factor tisular, que activa la cascada de la coagulación; del mismo modo, la activación de plaquetas favorecerá la síntesis de mediadores protrombóticos como el tromboxano A y las glicoproteínas IIb-IIIa. Otra teoría sobre su fisiopatología implica el daño oxidativo por el endotelio vascular por la reacción de los anticuerpos anticardiolipina con el LDL oxidado.
Sin embargo, es la activación de la cascada del complemento la que resulta relevante en las manifestaciones obstétricas, por insuficiencia placentaria, trombosis intraplacentaria o bien por placentación inefectiva.
Hipótesis del segundo hit:
Esta teoría defiende que debe existir un factor determinante que desencadene la enfermedad en presencia de los aFL, puesto que, como revelan los datos epidemiológicos, no todas las personas en las que se determinan estos anticuerpos se manifiesta la sintomatología del síndrome. Se consideran posibles factores desencadenantes los siguientes: infección, traumatismo, cirugía, inmovilización, embarazo o fármacos anticonceptivos, aunque no siempre son identificables.
CLÍNICA
Es una enfermedad sistémica, con unas manifestaciones clínicas heterogéneas; siendo las principales formas de presentación las trombosis arteriales o venosas y las manifestaciones obstétricas, como se ha comentado previamente. Si bien, tiende a conervar el patrón inicial de presentación, de forma que el segundo evento que ocurre suele ser del mismo tipo que el primero.
-Trombosis arteriales o venosas: afecta a vasos de cualquier calibre y localización, aunque la forma más frecuente es la TVP de miembros inferiores, pudiendo desarrollarse un tromboembolismo pulmonar e hipertensión pulmonar secundaria.
A nivel arterial, la afectación más frecuente suele ocurrir a nivel cerebral. Aunque localizaciones atípicas generan otros cuadros clínicos: síndrome coronario agudo, trombosis hepática arterial o venosa, isquemia portal, mesentérica o esplénica, e insuficiencia adrenal o pancreática secundarias.
La recurrencia de fenómenos trombóticos es frecuente, con una tasa de recurrencia en pacientes no tratados entre el 19-29%/año.
-Manifestaciones obstétricas: lo más frecuente son los abortos de repetición, frecuentemente de menos de 10 semanas de gestación; asociados o no a otros efectos adversos para la madre y el feto, como son las muertes fetales, CIR, preeclampsia o partos prematuros.
-Manifestaciones hematológicas: un 30% de los pacientes presentan trombocitopenia, pero raramente requiere tratamiento. Si asocia anemia hemolítica lo denominamos Síndrome de Evans. También puede aparecer Púrpura Trombocitopénica Idiopática como primera manifestación, por lo que en estos casos es recomendable medir los niveles de anticuerpos aFL.
-Manifestaciones cutáneas: La más característica es la lívedo reticularis, que está presente en un 25% de los pacientes, siendo un factor de riesgo independiente para la trombosis arterial. Otras menos frecuentes son las úlceras, fenómeno de Raynaud, tromboflebitis, hemorragias en astilla, púrpura y equimosis.
Ilustración 2. Lívedo Reticularis
-Otras: a nivel cardiaco encontramos enfermedad arterial coronaria trombótica junto a anomalías valvulares como manifestaciones más importantes; a nivel pulmonar puede aparecer hemorragia intraalveolar, síndrome de distrés respiratorio agudo y la alveolitis fibrosante. Las migrañas, epilepsia, corea… son manifestaciones neurológicas que ocasionalmente se asocian al síndrome antifosfolípido. La hipertensión arterial suele estar siempre presente y además puede ser el único signo clínico de nefropatía, aunque pueden desarrollar insuficiencia renal. A nivel ocular lo más frecuente es la afectación del segmento posterior, por isquemias e infartos en vasos retinianos.
SÍNDROME ANTIFOSFOLÍPIDO CATASTRÓFICO
El también llamado síndrome de Asherson se caracteriza por la aparición simultánea de trombosis en vasos de múltiples órganos, junto con microangiopatía, lo que provoca un fallo multiorgánico agudo. Es un proceso poco frecuente (<1% de los pacientes con SAF), pero con una alta mortalidad sin tratamiento (50%). En la siguiente tabla se adjuntan los criterios de clasificación:
Tabla 1. Criterios de clasificación del SAF catastrófico.
Tomado de: [1]
DIAGNÓSTICO
Es importante tener una sospecha clínica y realizar un diagnóstico precoz para disminuir la morbimortalidad del paciente.
Por su gran variabilidad clínica es una entidad infradiagnosticada; así que debemos plantear la posibilidad de estar ante un SAF si nuestro paciente:
-Ha sufrido una o más trombosis arterial o venosa, sin causa justificada, sobretodo en pacientes jóvenes.-Uno o más problemas como los comentados previamente durante el embarazo.-Trombocitopenia de causa desconocida, o alteración analítica en los tiempos de coagulación.-Diagnóstico previo de LES con aparición de sintomatología compatible.
Para realizar el diagnóstico debemos realizar 3 pruebas de laboratorio:
-ACL: mide la capacidad de los anticuerpos aFL de producir alteraciones in vitro en las pruebas de coagulación en las que intervienen los fosfolípidos (APTT, tiempo de veneno de víbora de Rusell diluido, tiempo de coagulación con caolín, o tiempo de protrombina…)-Anticuerpos anti-B2GPI-Anticuerpos anti-Cardiolipina
Los anticuerpos aFL pueden predecir el riesgo de aparición de complicaciones clínicas, que aumenta cuanto mayor es el número de anticuerpos involucrados.
Diagnóstico diferencial
El diagnóstico diferencial se puede realizar con otras enfermedades autoinmunes, que también podrían contar con la presencia de anticuerpos aFL. Ademas hay que tener en cuenta: trombofilias adquiridas, neoplasias y síndromes mieloproliferativos crónicos, enfermedad ateroesclerótica o hiperlipidemias, síndrome nefrótico, anticonceptivos orales, síndromes microangiopáticos, trombocitopenia inducida por heparina, coagulación intravascular diseminada o síndrome de Behçet.
En caso de manifestaciones obstétricas hay que descartar desequilibrios hormonales, coagulopatías, enfermedades autoinmunes y otras enfermedades crónicas inflamatorias, infecciones, intoxicaciones o abuso de drogas, anomalías genéticas fetales o en el cariotipo de los progenitores.
TRATAMIENTO
Es importante un tratamiento precoz para prevenir complicaciones y recurrencia de eventos trombóticos, aunque de forma rutinaria se puede hacer de manera ambulatoria, los cuadros más graves obligan a hospitalizar al paciente. El tratamiento se pautará de manera individual según el estado clínico y su historial médico previo, eliminando los factores de riesgo de trombosis y cardiovasculares. Y en aquellos que vayan a recibir tratamiento antiagregante y angicoagulante hay que valorar el riesgo de sangrado antes de iniciarlo.
-Tromboprofilaxis primaria: la introducción de tratamiento profiláctico en presencia de anticuerpos aFL, pero sin trombosis previa es controvertida, aunque siempre habrá que pautarlo si el paciente requiere hospitalización o cirugía. En los casos en los que hay una enfermedad autoinmune asociada hay que valorar especialmente el riesgo de trombosis, añadiendo Ácido Acetilsalicílico en casos de alto riesgo (LAC positivo, o triple positividad).
-Tromboprofilaxis secundaria: la recurrencia de fenómenos trombóticos justifica la anticoagulación de manera indefinida; teniendo en cuenta que si ha sido una trombosis venosa se anticoagulará de por vida, manteniendo un INR entre 2-3, con antagonistas de la vitamina K. En caso de trombosis arterial, se pauta anticoagulación crónica con un INR entre 3-4 en combinación con antiagregación con AAS. En aquellos pacientes con intolerancia o alergia a antagonistas de la vitamina K, se podrá considerar el uso de NACOS (nuevos anticoagulantes orales).
-Síndrome obstétrico: lo fundamental es instaurar el tratamiento en el momento más adecuado; estudios demuestran que tratar a mujeres con SAF aumenta la tasa de embarazos a término sin complicaciones, de forma que las indicaciones son las siguientes:
*Antecedentes obstétricos: AAS en dosis baja con Heparina de bajo peso molecular.*Antecedentes trombóticos: se sustituye la Warfarina por HBPM en dosis terapéuticas cuando se confirme el embarazo.
-Síndrome antifosfolípido catastrófico: las guías recomiendan anticoagulación plena con Heparina no fraccionada y glucocorticoides en dosis altas. Añadiendo Ciclofosfamida en casos de asociación con LES. Una segunda línea de tratamiento es el recambio plasmátco o las globulinas intravenosas, Rituximab y otros fármacos biológicos.
BIBLIOGRAFÍA
1.- Sobrino Grande, C.; Villalobos Sánchez, L.; Valero Expósito, M.; García Villanueva, M.J. (2017). Síndrome antifosfolípido. Medicine - Programa de Formación Médica Continuada Acreditado, 12(27), 1551–1559. doi:10.1016/j.med.2017.02.001
2.- Pacheco, R. A. (2009). Síndrome antifosfolípido. Revista Médica de Costa Rica y Centroamérica, 66(589), 313-317.
3.- Correa, A., Valderrama, O., Angel, R., Sáez, J., & Villablanca, E. (2002). Síndrome antifosfolípidos y embarazo. Revista chilena de obstetricia y ginecología, 67(3), 196-202.
4.- Alonso Santor, J. E., Inglada Galiana, L., & Pérez Paredes, G. (2007, May). Síndrome antifosfolípido: estado actual. In : Anales de medicina interna (Vol. 24, No. 5, pp. 242-248). Arán Ediciones, SL.


