Autora : María R. C.
4º Curso Medicina grupo "A" (curso 2021-2022)
Código de trabajo : 2101-MRC
INTRODUCCIÓN
Los recientes descubrimientos acerca de la relación entre el sistema inmunitario y la aparición de células cancerosas han permitido desarrollar las llamadas inmunoterapias. Tras el desarrollo de tratamientos con anticuerpos monoclonales, para restaurar funciones defensivas adormecidas por citoquinas del microambiente tumoral (como anti-PDL1 o anti CTLA 4) un nuevo marco de actuación está emergiendo: las terapias celulares. Concretamente las de células T, que responden a la interacción de sus receptores con antígenos concretos de las células diana. La investigación de las vías moleculares oncogénicas y de los marcadores tumorales de las últimas décadas asienta la base teórica que ha permitido este tipo de abordajes: los linfocitos T del propio paciente serán los que ataquen su tumor, tras haber sido extraídos y reprogramados genéticamente para presentar los receptores que se unan a los antígenos tumorales adecuados.
En este proceso intervienen por un lado los linfocitos T CD8, también llamados citotóxicos, que tras reconocer y unir sus receptores (de ahora en adelante, TCR) a su antígeno correspondiente, liberan sustancias citotóxicas que terminan con la diana, ya sea esta una célula infectada por un microorganismo, o en este caso, una célula tumoral que presentan moléculas específicas en su membrana. Por otro lado, son asimismo importantes los linfocitos T CD4, también llamados colaboradores, que tras unir su TCR a su antígeno específico, son activados para comenzar a liberar citocinas que a su vez favorecerán las respuestas de otras células inmunitarias.
En este proceso intervienen por un lado los linfocitos T CD8, también llamados citotóxicos, que tras reconocer y unir sus receptores (de ahora en adelante, TCR) a su antígeno correspondiente, liberan sustancias citotóxicas que terminan con la diana, ya sea esta una célula infectada por un microorganismo, o en este caso, una célula tumoral que presentan moléculas específicas en su membrana. Por otro lado, son asimismo importantes los linfocitos T CD4, también llamados colaboradores, que tras unir su TCR a su antígeno específico, son activados para comenzar a liberar citocinas que a su vez favorecerán las respuestas de otras células inmunitarias.
ORIGEN Y PASOS INICIALES
Antes incluso de que fuera posible la reprogramación genética de las células T, se sabía que estas podían eliminar células tumorales. Esta era la razón por la que en trasplantes de médula alogénicos se presenciaba la eliminación de células tumorales hematológicas: las linfocitos del donante no presentaban el mismo complejo de histocompatibilidad (MHC), así que reconocían el del receptor como extraños y terminaban con sus células. El problema, es que no solo las células tumorales presentaban su MHC particular, sino todas las del organismo, lo que daba lugar a la enfermedad del injerto contra el receptor. Se necesitaba una respuesta específica contra las células tumorales.
Hubo varios enfoques. Se intentó eliminar los linfocitos del donante, para solo trasplantar las células hematopoyéticas, introduciendo un gen suicida en los mismos a través de un retrovirus, pero esto reducía la respuesta antitumoral. Se buscó también potenciar la replicación de aquellos linfocitos de una muestra que interaccionaban más intensamente con antígenos de tumores como melanomas, mediante la administración de IL-2, pero la respuesta in vivo no resultaba la esperada. Finalmente, el desarrollo de técnicas de secuenciación masiva, de cultivos celulares y de vectores de introducción de genes concretos ha permitido utilizar las propias células del paciente.
Hubo varios enfoques. Se intentó eliminar los linfocitos del donante, para solo trasplantar las células hematopoyéticas, introduciendo un gen suicida en los mismos a través de un retrovirus, pero esto reducía la respuesta antitumoral. Se buscó también potenciar la replicación de aquellos linfocitos de una muestra que interaccionaban más intensamente con antígenos de tumores como melanomas, mediante la administración de IL-2, pero la respuesta in vivo no resultaba la esperada. Finalmente, el desarrollo de técnicas de secuenciación masiva, de cultivos celulares y de vectores de introducción de genes concretos ha permitido utilizar las propias células del paciente.
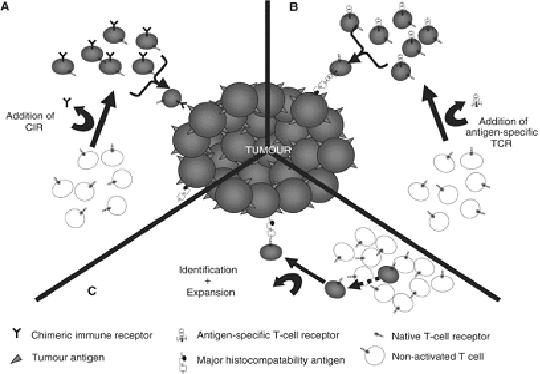
Ilustración 1: Mansoor et al, British Jounal of Cancer(2005). En este esquema vemos las diferentes estrategias mencionadas: A) células T con CIR B) células T tras introducción de gen por retrovirus C) células T específicas encontradas tras identificación de aquellas específicas y su expansión en cultivos
La posibilidad de introducir y fomentar la traducción de genes de las moléculas alfa y beta del TCR en la célula de un paciente ya no suponía un problema gracias a los vectores retrovirales. No obstante, seguía existiendo la limitación de no conocer suficientes genes de TCR que interaccionaran con tumores, salvo los que se habían descubierto por azar en muestras de linfocitos de donantes. Así, se consiguieron secuenciar genes TCR contra antígenos como MDM-2, presente en varios tumores, o WT-1, característico de algunas leucemias. Se necesitaba disponer de más genes de TCR para aumentar la oferta terapéutica, pero encontrar linfocitos T específicos de antígenos tumorales no era tan sencillo como conseguir anticuerpos monoclonales.
Es por esto que los receptores inmunes quiméricos (CIR) permitieron en gran medida el desarrollo de estas terapias. Estos consistían en la fusión de un anticuerpo monoclonal (específico de un antígeno tumoral) con una cadena beta de TCR. La unión del anticuerpo a su antígeno activaba el receptor y la liberación de los gránulos citotóxicos, y lo que es más importante, esto era posible sin la necesidad de presentar los antígenos por parte de MHC. Teniendo en cuenta que muchos tumores ven disminuida la cantidad de MHC expresado, los receptores inmunes quiméricos cobraban aún más importancia. Desde entonces, se disponen de células T con CIR específicos de CD20, CD30, CEA, ErbB-2…(1)
Es por esto que los receptores inmunes quiméricos (CIR) permitieron en gran medida el desarrollo de estas terapias. Estos consistían en la fusión de un anticuerpo monoclonal (específico de un antígeno tumoral) con una cadena beta de TCR. La unión del anticuerpo a su antígeno activaba el receptor y la liberación de los gránulos citotóxicos, y lo que es más importante, esto era posible sin la necesidad de presentar los antígenos por parte de MHC. Teniendo en cuenta que muchos tumores ven disminuida la cantidad de MHC expresado, los receptores inmunes quiméricos cobraban aún más importancia. Desde entonces, se disponen de células T con CIR específicos de CD20, CD30, CEA, ErbB-2…(1)

Ilustración 2: Hughes-Parry et al. International Journal of Molecular Sciencies (2020). En este esquema vemos como se conformaría el receptor tipo CIR, con la fracción variable de un anticuerpo monoclonal unido al dominio transmembrana de la cadena beta y al CD3ζ.(2)
TERAPIAS CAR-T PARA TUMORES DE CÉLULAS B
Los primeros CIR obtenidos fueron contra el antígeno CD19, presente en los linfocitos B, lo que ha permitido que las primeras terapias de uso clínico hayan sido para tumores de este tipo de células. Actualmente también se dispone de tratamientos contra mielomas (antígeno BCMA), y se pretende ampliar el abanico de oferta terapéutica, sin embargo, los ensayos clínicos de terapias para tumores sólidos de momento muestran una eficacia limitada y gran toxicidad. Esto último se puede deber a la dificultad de encontrar antígenos específicos, como mencionábamos anteriormente, así como a la pobre infiltración de los linfocitos T reprogramados y el microambiente inmunosupresor del tumor.
Es por esto que la investigación centrada en los tumores hematológicos ha ido dando más frutos. Actualmente los CIR se han perfeccionado, obteniéndose así los llamados de “segunda generación”, en los que se fusiona solo una cadena de la región variable del anticuerpo monoclonal con la cadena beta del receptor TCR, con la novedad de estar esta a su vez conectada a dominios de receptores coestimuladores. Estos CIR han demostrado una mayor eficacia a la hora de producir citocinas proinflamatorias y favorecer la citotoxicidad contra el tumor. (3)
Es por esto que la investigación centrada en los tumores hematológicos ha ido dando más frutos. Actualmente los CIR se han perfeccionado, obteniéndose así los llamados de “segunda generación”, en los que se fusiona solo una cadena de la región variable del anticuerpo monoclonal con la cadena beta del receptor TCR, con la novedad de estar esta a su vez conectada a dominios de receptores coestimuladores. Estos CIR han demostrado una mayor eficacia a la hora de producir citocinas proinflamatorias y favorecer la citotoxicidad contra el tumor. (3)
La verdadera limitación que se han encontrado este tipo de terapias ha sido su alto coste. La aprobación en 2018 de la primera terapia CAR-T de uso comercial, “Kimria” del laboratorio Novartis, no se vio exenta de polémica. Su precio de 320.000€ en Europa (excesivamente caro, teniendo en cuenta que según investigadores de este ámbito, el precio de fabricación podía rondar los 20.000 €), impidió que se pudiera a aplicar a todos los pacientes indicados (pacientes infantiles de leucemia linfoblástica aguda en recaída y pacientes adultos en recaída de linfoma B difuso de célula grande). Tras diversos litigios judiciales por problemas de trasparencia, se consiguió anular parcialmente la patente, si bien el tratamiento sigue sin ser precisamente económico. Es por esto que el desarrollo desde el ámbito público, desde la investigación básica hasta la cama del paciente, es un rayo de esperanza para todos estos pacientes que ven sus posibilidades de tratamiento tan limitadas. (4)(5)(6)(7)
EL PROYECTO ARI EN BARCELONA: CAR-T ARI-0001
El primer y único proyecto de terapia CAR-T en España fue desarrollado en Barcelona. Es a su vez el primero enteramente público, si bien hay que agradecer su impulso principal: el “Proyecto Ari”, organización benéfica fundada por Ari Benedé, una chica diagnosticada con leucemia linfoblástica aguda que desgraciadamente falleció en 2016. Al trabajo de ella y de su madre se debe gran parte de la financiación.

Ilustración 3 : Ariana Benedé (1998-2016)
Imagen obtenida en internet, sin créditos de autor, de probable dominio público.
En este proyecto se utilizó un anticuerpo anti-CD19 (A3B1) para unirlo con la cadena beta en el CIR. Este había sido desarrollado 30 años antes también en el mismo hospital, el Clinic de Barcelona, para utilizarse como anticuerpo monoclonal. Para ello, primero volvieron a demostrar la especificidad del anticuerpo, y tras eso, la citotoxicidad por parte de los linfocito T reprogramados, gracias a estudios de citometría de flujo que indicaban que, tras el cultivo conjunto, las células CD19+, así como la medición de citocinas producidas mediante ELISA, concretamente, los niveles de TNF alfa y IFN gamma se vieron aumentados.
Tras demostrar su efectividad in vitro, se procedió a los ensayos con ratones, donde con biopsias se confirmó no solo la no progresión de la enfermedad, sino también la eliminación de las células CD19+. Se comparó la efectividad con otras células CAR-T y no hubo diferencias significativas. Solo quedaba poder replicar a gran escala los lentivirus que introdujeran los genes del CIR, y después cultivar las células. Gracias a maquinaria altamente especializada, el biorreactor cerrado semiautomático de CliniMACS Prodigy, de Miltenyi Biotec se consiguieron cantidades adecuadas, que si bien no a un ritmo industrial, permitía comenzar los ensayos clínicos, y tras sus prometedores resultados, empezar a tratar a todos aquellos pacientes que cumplieran con los criterios de inclusión. (8)
Para realizar los ensayos clínicos que permitieran la aprobación como medicamento por parte del ministerio de sanidad, se incluyeron a 28 pacientes tanto pediátricos como adultos, del Hospital Clinic y del Hospital Sant Joan de Déu. Esto es importante porque los otros tratamientos comerciales (Kimria y Yescarta) no estaban indicados para pacientes adultos mayores de 25 años.
En la fase I de los ensayos clínicos, mediante aféresis se extraía sangre de los pacientes, se eliminaban los eritrocitos y plaquetas gracias a centrifugación y se seleccionaban los linfocitos T con bandas magnéticas. Se comenzaban los cultivos como se ha mencionado previamente, durante 7-10 días, donde las células se expandían con IL-7 y IL-15, y después se criopreservaban hasta la infusión. El objetivo era tener 2 dosis por paciente, de 1 millón de células por kilo para pacientes de leucemia linfoblastica aguda y crónica y 5 millones de células por kilo para aquellos pacientes de linfoma no hodgkin. Para el control de calidad, se realizaban PCR para asegurar la correcta inclusión del gen, medición de citoquinas… Se pudo demostrar que todos los productos eran citotóxicos contra células CD-19+, que se producían citoquinas proinflamatorias, pero también, que la expansión era menor en las células de pacientes que en células de donantes sanos. También se descubrió que una proporción de las células T eran capaces generar memoria inmunitaria en los pacientes. Se demostraba que conseguir el tratamiento con ese tipo de biorreactores y que funcionaran en pacientes era factible. (3)
Finalmente, los resultados del ensayo clínico permitieron la aprobación del fármaco por la AEMPS. Los pacientes recibieron ciclofosfamida, fludarabina y después entre 0,4 y 5 millones de células ARI-0001 por kg. Al principio se dio una sola dosis, y después se dividió en tres dosis del 10%, el 30% y el 60% para prevenir una de las peores complicaciones, el síndrome de liberación de citoquinas(SLC). De los 58 pacientes incluidos en el estudio, 47 recibieron la terapia, 38 de los cuales presentaban LLA, 7 linfoma no Hodgkin y el restante LLC. En los pacientes con LLA, un 13,2 % presentó SLC, que fue tratada con tocilizumab. Un 2,6% de los pacientes presentaron neurotoxicidad, y la mortalidad relacionada con el procedimiento fue de 7,9% ras el día 100, sin ninguna otra muerte tras espaciar la dosis. El porcentaje de remisión completa en el día 100 fue de un 71,1%, y la supervivencia sin progresión fue de un 47% al año. La supervivencia en general fue de un 68,6% en definitiva, cifras similares a las de las otras dos soluciones comerciales. (3)(9)(10)(11)
COMPLICACIONES DE LA TERAPIA CAR-T
Las siguientes complicaciones son las que se describen en la ficha técnica de CAR-T ARI-0001, que son comunes a otros productos comerciales.
· Reacciones a la infusión: por lo general leves, que nunca han llevado a la interrupción temporal o a la retirada del tratamiento. Por este motivo se administra premedicación 30 minutos antes de la infusión (paracetamol, dexclorfeniramina) Pueden ser: eventos cardíacos, escalofríos, disnea, fatiga, hipertensión repentina, hipotensión, náuseas, dolor, fiebre, erupción cutánea y urticaria.
· Síndrome de lisis tumoral (SLT): Para prevenirlo se recomienda la administración oral de alopurinol e hidratación oral abundante (al menos 2 litros/día) a partir del día de la infusión Se deben monitorizar los signos y síntomas de SLT (creatinemia, arritmias por la liberación de potasio, convulsiones…)
· Reacciones a la infusión: por lo general leves, que nunca han llevado a la interrupción temporal o a la retirada del tratamiento. Por este motivo se administra premedicación 30 minutos antes de la infusión (paracetamol, dexclorfeniramina) Pueden ser: eventos cardíacos, escalofríos, disnea, fatiga, hipertensión repentina, hipotensión, náuseas, dolor, fiebre, erupción cutánea y urticaria.
· Síndrome de lisis tumoral (SLT): Para prevenirlo se recomienda la administración oral de alopurinol e hidratación oral abundante (al menos 2 litros/día) a partir del día de la infusión Se deben monitorizar los signos y síntomas de SLT (creatinemia, arritmias por la liberación de potasio, convulsiones…)
· Síndrome de liberación de citocinas (SLC): se observó SLC en un 55% de los pacientes y en 13,2% fue de grado III o superior (fiebre persistente, de más de 38ºC, asociada a: hipoxia que requiere ventimask o cánula nasal de alto flujo o hipotensión arterial que requiere la administración de un único vasopresor). Los síntomas del SLC podrían ser, aparte de los mencionados, escalofríos, mialgia, artralgias, náuseas, vómitos, diarrea, diaforesis, erupción, anorexia, fatiga, dolor de cabeza, disnea, taquipnea. Pueden observarse también alteraciones orgánicas como insuficiencia cardiaca y arritmia, insuficiencia renal y daño hepático acompañado de elevaciones de la AST, ALT o de la bilirrubina total. En algunos casos podría aparecer coagulación intravascular diseminada (CID), con bajos niveles de fibrinógeno, síndrome de fuga capilar, síndrome de activación macrofágica (SAM) y linfohistiocitosis hemofagocítica. Se ha de administrar un tratamiento profiláctico y terapéutico para las infecciones y se ha de garantizar la resolución completa de las infecciones existentes. Las infecciones también pueden ocurrir durante el SLC y podrían incrementar el riesgo de muerte. El síndrome de liberación de citoquinas debe manejarse administrando toicilizumab o corticoides dependiendo del grado de gravedad. Ante la aparición de hipotensión o hipoxia, se debe considerar el traslado a una Unidad de Cuidados Intensivos (UCI).
· Neurotoxicidad (ICANS): se detectaron alteraciones neurológicas en 16 pacientes con LLA, de las cuales 1 fue considerada como neurotoxicidad asociada a las células inmunoefectoras (ICANS) Puede tener diversas formas de presentación: temblor fino, digrafía, dificultad leve en el habla (afasia de expresión), déficit de atención, apraxia y letargo leve. La cefalea es frecuente pero inespecífica, mientras que la afasia de expresión es muy específica. Los síntomas pueden empeorar hasta afasia global, llegando incluso al mutismo y la acinesia. También pueden aparecer mioclonías o temblor grave, nistagmo, visión borrosa, disminución del nivel de conciencia, letargo, obnubilación, estupor o incluso coma. Son posibles las convulsiones, casi siempre después de afasia global. En casos raros las pruebas de imagen (RM) demuestran la aparición de edema cerebral, local o general, que puede ser de instauración muy rápida. Se trata también con tocilizumab o corticoides, incluso terapia osmótica en caso de edema cerebral.
· Otras complicaciones: aplasia de linfocitos B (si bien en esos pacientes no se produjeron más infecciones que en otro tipo de tratamientos), segundas neoplasias, … (12)(13)(14)
NUEVOS HORIZONTES
Está claro que las terapias CAR-T han supuesto una verdadera revolución de cara al tratamiento de pacientes oncológicos, principalmente hematológicos por el momento. Aunque actualmente su elevado coste y sus complicaciones hacen que sean vistas como último recurso para pacientes que no responden a terapias tradicionales, está claro que las futuras investigaciones supondrán grandes avances que permitirán su generalización
Si se sigue favoreciendo el desarrollo público que permita el acceso por parte de candidatos, y se tiene más información acerca del mecanismo fisiopatológico de las complicaciones mencionadas y su tratamiento, cada vez podrán ser más utilizadas.
Las líneas de investigación actuales señalan que los posibles proyectos se basarán en la creación de células CAR-T madre, que favorece su durabilidad y la total remisión de la enfermedad, así como células de cuarta generación que puedan actuar contra tumores sólidos como células T que contengan genes suicidas o CAR-T productoras de anticuerpos. (15)
Si se sigue favoreciendo el desarrollo público que permita el acceso por parte de candidatos, y se tiene más información acerca del mecanismo fisiopatológico de las complicaciones mencionadas y su tratamiento, cada vez podrán ser más utilizadas.
Las líneas de investigación actuales señalan que los posibles proyectos se basarán en la creación de células CAR-T madre, que favorece su durabilidad y la total remisión de la enfermedad, así como células de cuarta generación que puedan actuar contra tumores sólidos como células T que contengan genes suicidas o CAR-T productoras de anticuerpos. (15)
BIBLIOGRAFÍA
Además de las fuentes mencionadas, parte de los conocimientos para realizar este trabajo son los adquiridos en la asignatura de Inmunología de 2º.1. Mansoor W, Gilham DE, Thistlethwaite FC, Hawkins RE. Engineering T cells for cancer therapy. Br J Cancer [Internet]. 2005 Nov 4 [cited 2021 Sep 8];93(10):1085. Available from: /pmc/articles/PMC2361500
2. Hughes-Parry HE, Cross RS, Jenkins MR. The Evolving Protein Engineering in the Design of Chimeric Antigen Receptor T Cells. Int J Mol Sci 2020, Vol 21, Page 204 [Internet]. 2019 Dec 27 [cited 2021 Oct 20];21(1):204. Available from: https://www.mdpi.com/1422-0067/21/1/204/htm
3. Castella M, Caballero-Baños M, Ortiz-Maldonado V, González-Navarro EA, Suñé G, Antoñana-Vidósola A, et al. Point-Of-Care CAR T-Cell Production (ARI- 0001) Using a Closed Semi-automatic Bioreactor: Experience From an Academic Phase I Clinical Trial. Front Immunol. 2020;0:482
4. Los hospitales públicos pagan 307.200 euros por cada tratamiento personalizado contra la leucemia infantil [Internet]. [cited 2021 Sep 11]. Available from: https://civio.es/medicamentalia/2019/10/29/car-t-kymriah-yescarta- precios-novartis-gilead
5. Informe de Posicionamiento Terapéutico de tisagenlecleucel (Kymriah®) en el tratamiento de pacientes pediátricos y adultos hasta 25 años con leucemia linfoblástica aguda de células B refractaria, en recaída post-trasplante, o en segunda recaída o posterior; y de pacientes adultos con linfoma difuso de células grandes B recaído/refractario tras dos o más líneas de tratamiento sistémico. Ministerio de Sanidad. 2019 Feb 25. [cited 2021 Sep 8]. Available from: /https://www.aemps.gob.es/medicamentosUsoHumano/informesPublicos/docs/I PT-tisagenlecleucel-kymriah-LAL-LCGB.pdf
6. Novartis se enfrenta a la OCU por el precio Kymriah [Internet]. [cited 2021 Sep 11].Available from: https://www.redaccionmedica.com/secciones/industria/novartis-recurre-que-se- publiquen-los-criterios-de-financiacion-de-kymriah-8264
7. Victoria de Médicos del Mundo: Novartis retira su patente abusiva sobre un tratamiento contra el cáncer desmesuradamente caro | Médicos del Mundo [Internet]. [cited 2021 Sep 11]. Available from: https://www.medicosdelmundo.org/actualidad-y-publicaciones/noticias/victoria- de-medicos-del-mundo-novartis-retira-su-patente-abusiva
8. Castella M, Boronat A, Martín-Ibáñez R, Rodríguez V, Suñé G, Caballero M, et al. Development of a Novel Anti-CD19 Chimeric Antigen Receptor: A Paradigm for an Affordable CAR T Cell Production at Academic Institutions. Mol Ther - Methods Clin Dev [Internet]. 2019 Mar 15 [cited 2021 Sep 11];12:134–44. Available from: http://www.cell.com/article/S2329050118301220/fulltext
9. Juan DM, Delgado DJ, Calvo DG, Trias DE, Urbano-Ispizua PA. Is Hospital Exemption an alternative or a bridge to EMA for developing Academic CAR-T in Europe? Our experience with ARI0001. https://home.liebertpub.com/hum [Internet]. 2021 Sep 3 [cited 2021 Sep 11]; Available from: https://www.liebertpub.com/doi/abs/10.1089/hum.2021.168
10. Ortíz-Maldonado V, Rives S, Castellà M, Alonso-Saladrigues A, Benítez- Ribas D, Caballero-Baños M, et al. CART19-BE-01: A Multicenter Trial of ARI- 0001 Cell Therapy in Patients with CD19 + Relapsed/Refractory Malignancies. Mol Ther [Internet]. 2021 Feb 3 [cited 2021 Sep 11];29(2):636–44. Available from: https://pubmed.ncbi.nlm.nih.gov/33010231/
11. Castellà M, Boronat A, Martín-Ibáñez R, Rodríguez V, Suñé G, Caballero M, et al. Development of a Novel Anti-CD19 Chimeric Antigen Receptor: A Paradigm for an Affordable CAR T Cell Production at Academic Institutions. Mol Ther Methods Clin Dev [Internet]. 2018 Mar 15 [cited 2021 Sep 11];12:134–44. Available from: https://pubmed.ncbi.nlm.nih.gov/30623002/
12. Investigación clínica, terapias avanzadas [Internet]. [cited 2021 Sep 11]. Available from: https://www.aemps.gob.es/investigacionClinica/terapiasAvanzadas/docs/ARI- 0001_ficha-tecnica.pdf?x12870
13. Notario Dongil C, Fraga Fuentes M, Gómez Lluch M, Marcos de La Torre A, Andrés Navarro N, Valenzuela Gámez J, et al. CAR-T: luces y sombras. Rev la OFIL [Internet]. 2020 [cited 2021 Sep 11];30(4):329–33. Available from: https://scielo.isciii.es/scielo.php?script=sci_arttext&pid=S1699- 714X2020000400011&lng=es&nrm=iso&tlng=es
14. Yakoub-Agha I, Chabannon C, Bader P, Basak GW, Bonig H, Ciceri F, et al. Management of adults and children undergoing chimeric antigen receptor T-cell therapy: best practice recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE). Haematologica [Internet]. 2020 Jan 31 [cited 2021 Sep 11];105(2):297–316. Available from: https://pubmed.ncbi.nlm.nih.gov/31753925/ McLellan AD, Ali Hosseini Rad SM. Chimeric antigen receptor T cell persistence and memory cell formation. Immunol Cell Biol [Internet]. 2019 [cited 2021 Sep 11];97(7):664–74. Available from: https://pubmed.ncbi.nlm.nih.gov/31009109/